

AKAP150-dependent cooperative TRPV4 channel gating is central to endothelium-dependent vasodilation and is disrupted in hypertension
- PMID: 25005230
- PMCID: PMC4403000
- DOI: 10.1126/scisignal.2005052
AKAP150-dependent cooperative TRPV4 channel gating is central to endothelium-dependent vasodilation and is disrupted in hypertension
Abstract
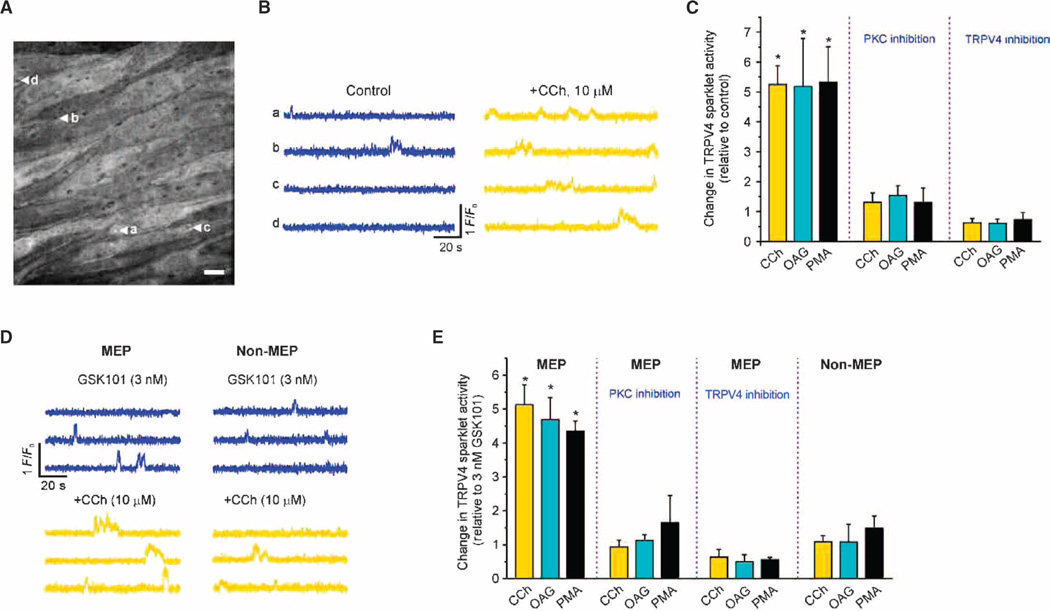
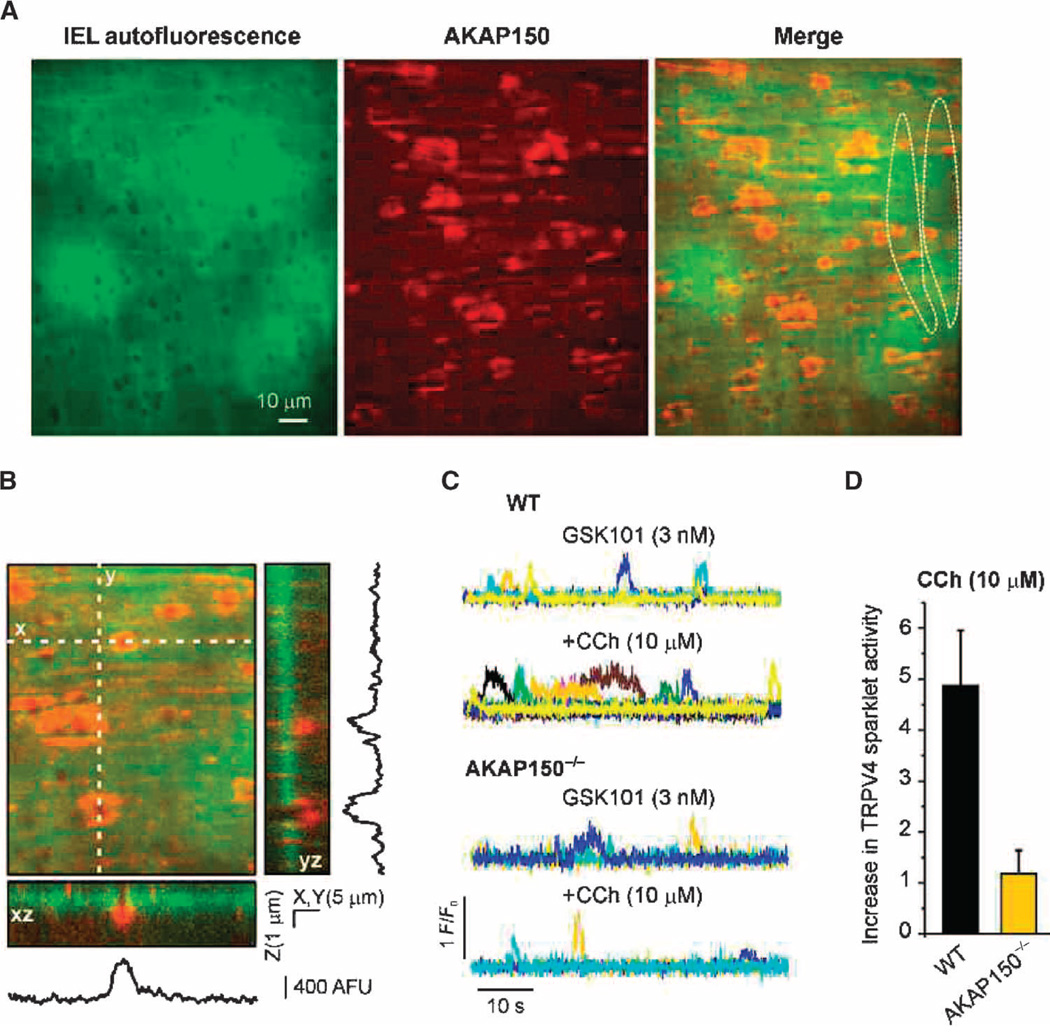
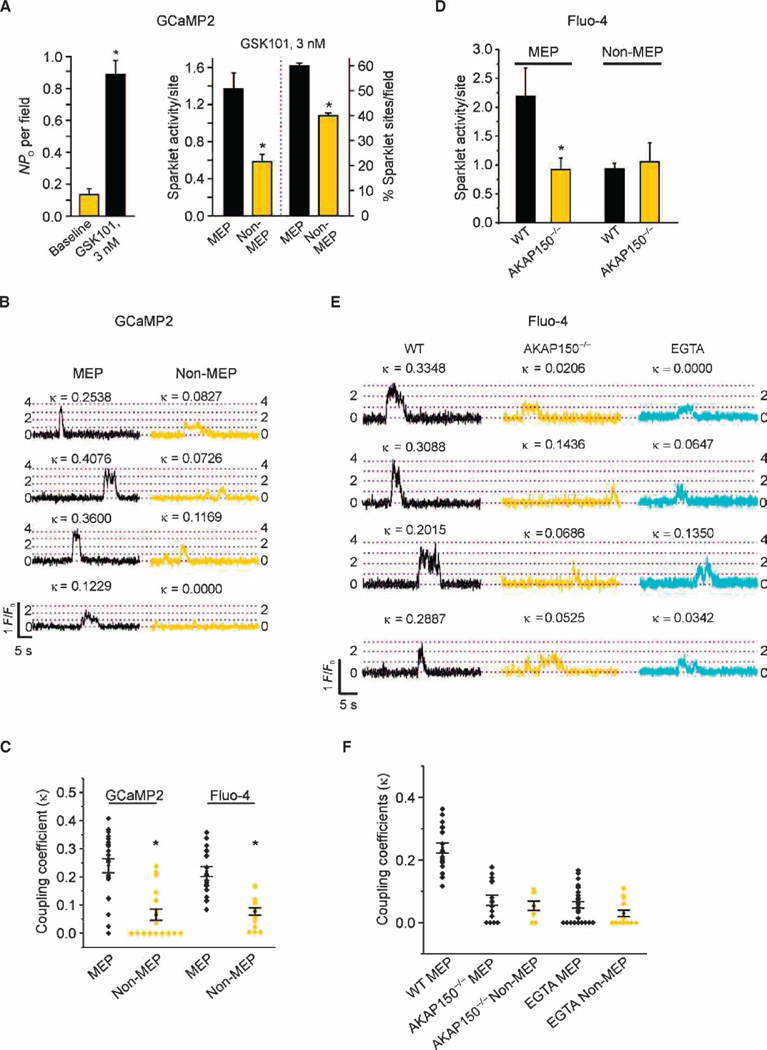
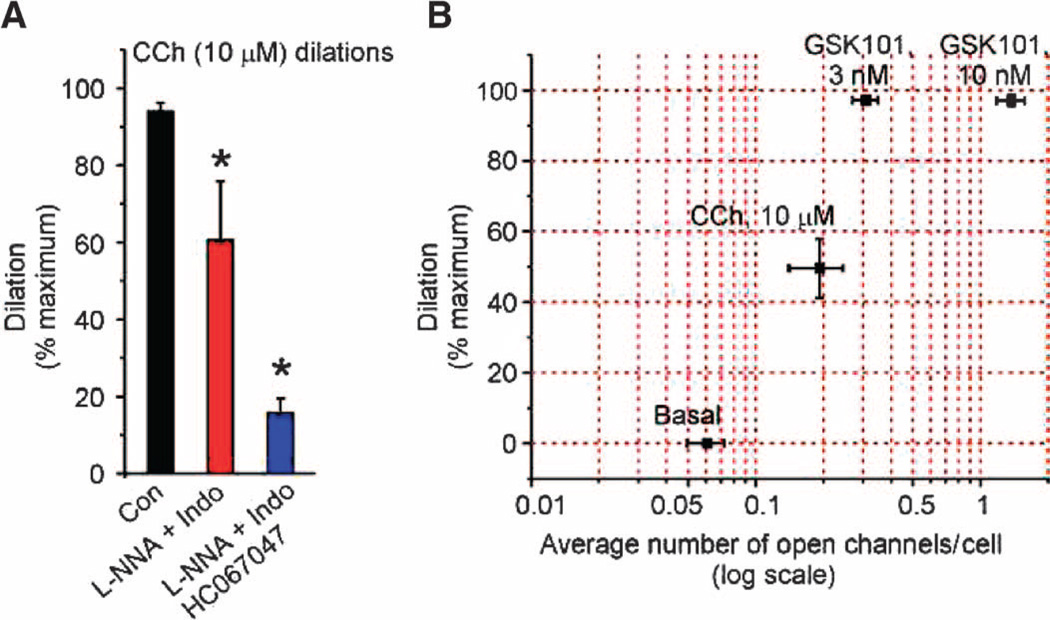
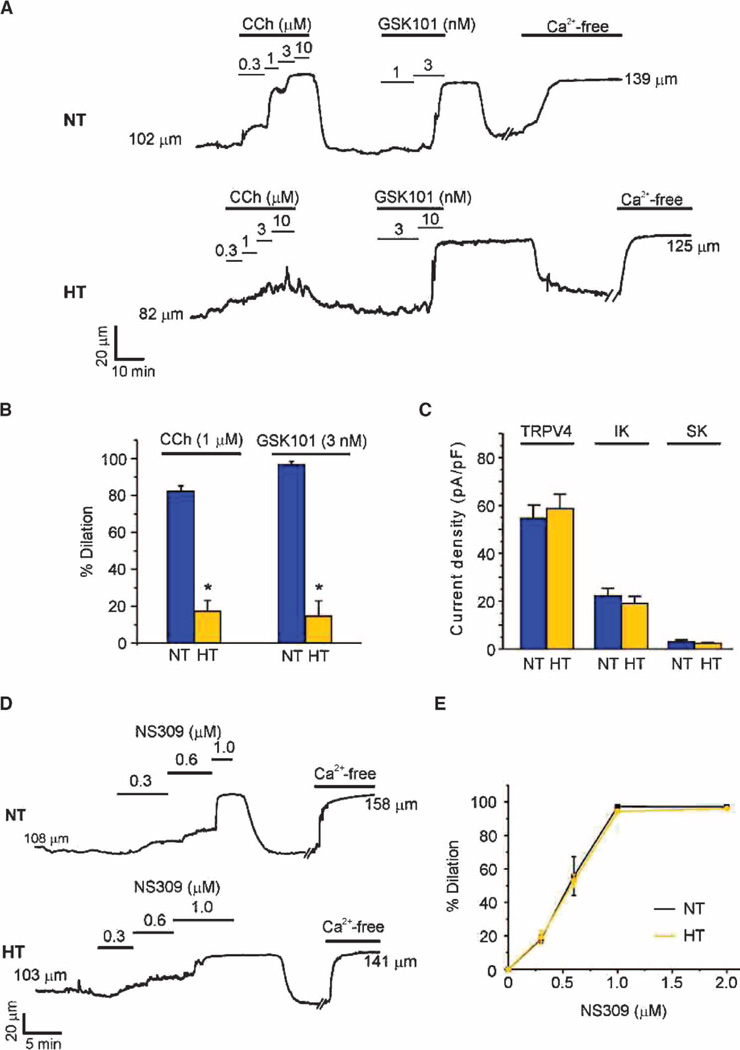
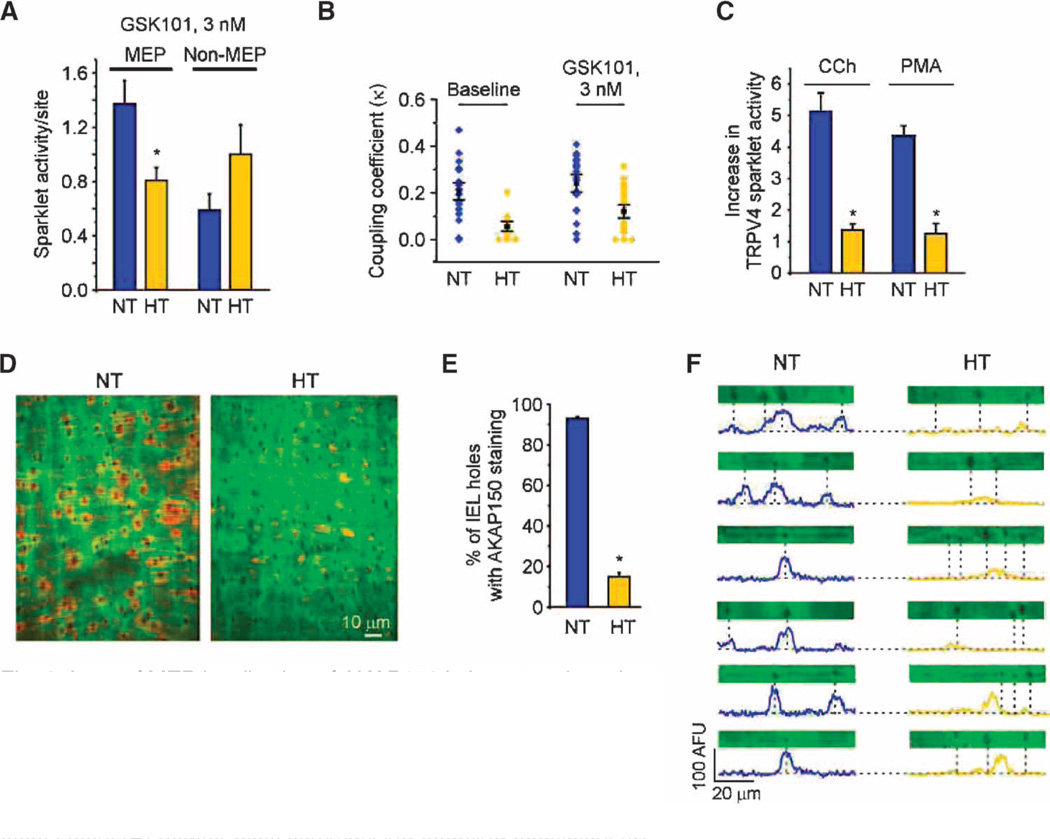
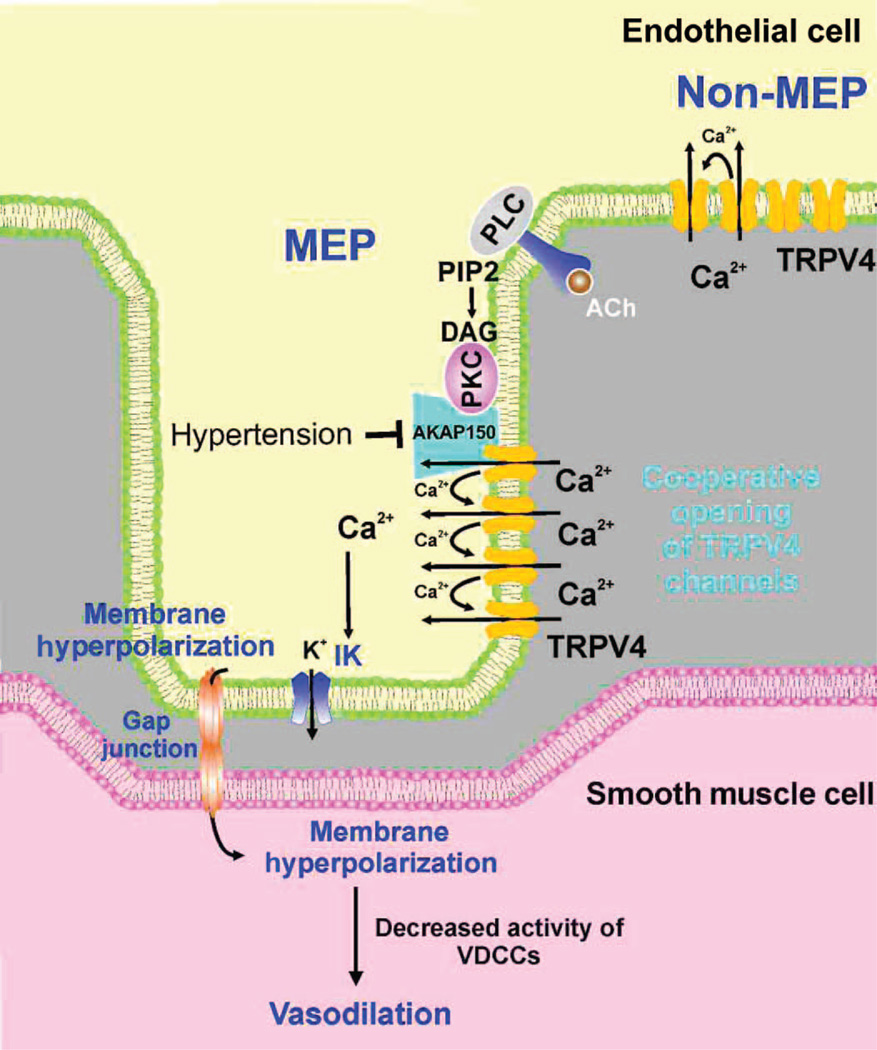
Endothelial cell dysfunction, characterized by a diminished response to endothelial cell-dependent vasodilators, is a hallmark of hypertension. TRPV4 channels play a major role in endothelial-dependent vasodilation, a function mediated by local Ca(2+) influx through clusters of functionally coupled TRPV4 channels rather than by a global increase in endothelial cell Ca(2+). We showed that stimulation of muscarinic acetylcholine receptors on endothelial cells of mouse arteries exclusively activated TRPV4 channels that were localized at myoendothelial projections (MEPs), specialized regions of endothelial cells that contact smooth muscle cells. Muscarinic receptor-mediated activation of TRPV4 depended on protein kinase C (PKC) and the PKC-anchoring protein AKAP150, which was concentrated at MEPs. Cooperative opening of clustered TRPV4 channels specifically amplified Ca(2+) influx at MEPs. Cooperativity of TRPV4 channels at non-MEP sites was much lower, and cooperativity at MEPs was greatly reduced by chelation of intracellular Ca(2+) or AKAP150 knockout, suggesting that Ca(2+) entering through adjacent channels underlies the AKAP150-dependent potentiation of TRPV4 activity. In a mouse model of angiotensin II-induced hypertension, MEP localization of AKAP150 was disrupted, muscarinic receptor stimulation did not activate TRPV4 channels, cooperativity among TRPV4 channels at MEPs was weaker, and vasodilation in response to muscarinic receptor stimulation was reduced. Thus, endothelial-dependent dilation of resistance arteries is enabled by MEP-localized AKAP150, which ensures the proximity of PKC to TRPV4 channels and the coupled channel gating necessary for efficient communication from endothelial to smooth muscle cells in arteries. Disruption of this molecular assembly may contribute to altered blood flow in hypertension.
Copyright © 2014, American Association for the Advancement of Science.
Figures
References
-
- Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. - PubMed
-
- Dora KA. Coordination of vasomotor responses by the endothelium. Circ. J. 2010;74:226–232. - PubMed
-
- Sandow SL, Senadheera S, Grayson TH, Welsh DG, Murphy TV. Calcium and endothelium-mediated vasodilator signaling. Adv. Exp. Med. Biol. 2012;740:811–831. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- GM086736/GM/NIGMS NIH HHS/United States
- 2-P20-RR-016435-06/RR/NCRR NIH HHS/United States
- K99 HL121484/HL/NHLBI NIH HHS/United States
- P20 RR016435/RR/NCRR NIH HHS/United States
- R01 GM086736/GM/NIGMS NIH HHS/United States
- R37 GM048231/GM/NIGMS NIH HHS/United States
- R01HL098243/HL/NHLBI NIH HHS/United States
- R37 DK053832/DK/NIDDK NIH HHS/United States
- 1P01HL095488/HL/NHLBI NIH HHS/United States
- R37DK053832/DK/NIDDK NIH HHS/United States
- R01 HL044455/HL/NHLBI NIH HHS/United States
- R37GM48231/GM/NIGMS NIH HHS/United States
- P01 HL095488/HL/NHLBI NIH HHS/United States
- R01 HL085870/HL/NHLBI NIH HHS/United States
- HL044455/HL/NHLBI NIH HHS/United States
- R01 DK053832/DK/NIDDK NIH HHS/United States
- R01 HL098243/HL/NHLBI NIH HHS/United States
- R00 HL121484/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
