

The role of the renin-angiotensin-aldosterone system in the pathobiology of pulmonary arterial hypertension (2013 Grover Conference series)
- PMID: 25006439
- PMCID: PMC4070776
- DOI: 10.1086/675984
The role of the renin-angiotensin-aldosterone system in the pathobiology of pulmonary arterial hypertension (2013 Grover Conference series)
Abstract
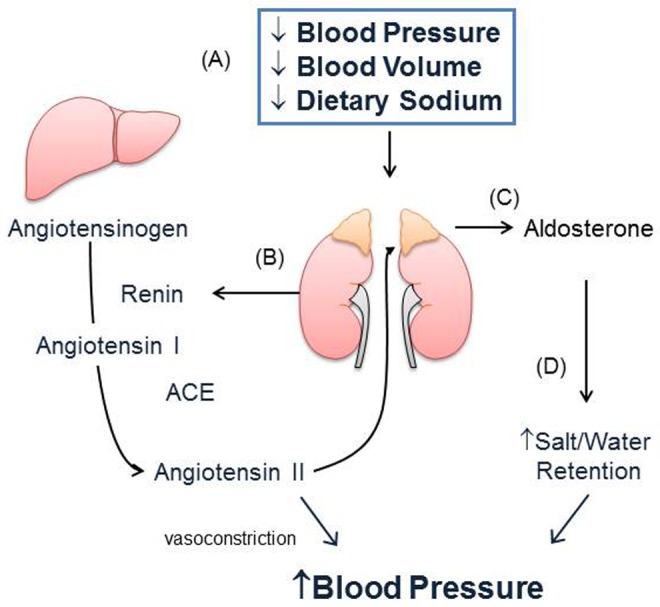
Pulmonary arterial hypertension (PAH) is associated with aberrant pulmonary vascular remodeling that leads to increased pulmonary artery pressure, pulmonary vascular resistance, and right ventricular dysfunction. There is now accumulating evidence that the renin-angiotensin-aldosterone system is activated and contributes to cardiopulmonary remodeling that occurs in PAH. Increased plasma and lung tissue levels of angiotensin and aldosterone have been detected in experimental models of PAH and shown to correlate with cardiopulmonary hemodynamics and pulmonary vascular remodeling. These processes are abrogated by treatment with angiotensin receptor or mineralocorticoid receptor antagonists. At a cellular level, angiotensin and aldosterone activate oxidant stress signaling pathways that decrease levels of bioavailable nitric oxide, increase inflammation, and promote cell proliferation, migration, extracellular matrix remodeling, and fibrosis. Clinically, enhanced renin-angiotensin activity and elevated levels of aldosterone have been detected in patients with PAH, which suggests a role for angiotensin and mineralocorticoid receptor antagonists in the treatment of PAH. This review will examine the current evidence linking renin-angiotensin-aldosterone system activation to PAH with an emphasis on the cellular and molecular mechanisms that are modulated by aldosterone and may be of importance for the pathobiology of PAH.
Keywords: aldosterone; angiotensin II; mineralocorticoid receptor; pulmonary arterial hypertension; spironolactone.
Figures


Similar articles
-
Aldosterone inactivates the endothelin-B receptor via a cysteinyl thiol redox switch to decrease pulmonary endothelial nitric oxide levels and modulate pulmonary arterial hypertension.Circulation. 2012 Aug 21;126(8):963-74. doi: 10.1161/CIRCULATIONAHA.112.094722. Epub 2012 Jul 11. Circulation. 2012. PMID: 22787113 Free PMC article.
-
Dysregulated renin-angiotensin-aldosterone system contributes to pulmonary arterial hypertension.Am J Respir Crit Care Med. 2012 Oct 15;186(8):780-9. doi: 10.1164/rccm.201203-0411OC. Epub 2012 Aug 2. Am J Respir Crit Care Med. 2012. PMID: 22859525 Free PMC article.
-
Involvement of the bone morphogenetic protein system in endothelin- and aldosterone-induced cell proliferation of pulmonary arterial smooth muscle cells isolated from human patients with pulmonary arterial hypertension.Hypertens Res. 2010 May;33(5):435-45. doi: 10.1038/hr.2010.16. Epub 2010 Feb 26. Hypertens Res. 2010. PMID: 20186146
-
Aldosterone and Mineralocorticoid Receptor Antagonists on Pulmonary Hypertension and Right Ventricular Failure: A Review.Curr Pharm Des. 2020;26(31):3862-3870. doi: 10.2174/1381612826666200523171137. Curr Pharm Des. 2020. PMID: 32445449 Review.
-
Neurohormonal activation and pharmacological inhibition in pulmonary arterial hypertension and related right ventricular failure.Heart Fail Rev. 2016 Sep;21(5):539-47. doi: 10.1007/s10741-016-9566-3. Heart Fail Rev. 2016. PMID: 27206576 Review.
Cited by
-
The efficacy and safety of Sacubitril/Valsartan on pulmonary hypertension in hemodialysis patients.Front Med (Lausanne). 2022 Nov 29;9:1055330. doi: 10.3389/fmed.2022.1055330. eCollection 2022. Front Med (Lausanne). 2022. PMID: 36523777 Free PMC article.
-
Pulmonary hypertension: diagnostic and therapeutic challenges.Ther Clin Risk Manag. 2015 Aug 17;11:1221-33. doi: 10.2147/TCRM.S74881. eCollection 2015. Ther Clin Risk Manag. 2015. PMID: 26316767 Free PMC article. Review.
-
Angiotensin II type 1 receptor mediates pulmonary hypertension and right ventricular remodeling induced by inhaled nicotine.Am J Physiol Heart Circ Physiol. 2021 Apr 1;320(4):H1526-H1534. doi: 10.1152/ajpheart.00883.2020. Epub 2021 Feb 12. Am J Physiol Heart Circ Physiol. 2021. PMID: 33577434 Free PMC article.
-
Pathogenic Mechanisms of Pulmonary Arterial Hypertension: Homeostasis Imbalance of Endothelium-Derived Relaxing and Contracting Factors.JACC Asia. 2022 Dec 15;2(7):787-802. doi: 10.1016/j.jacasi.2022.09.010. eCollection 2022 Dec. JACC Asia. 2022. PMID: 36713766 Free PMC article. Review.
-
Lower Plasma Melatonin Levels Predict Worse Long-Term Survival in Pulmonary Arterial Hypertension.J Clin Med. 2020 Apr 25;9(5):1248. doi: 10.3390/jcm9051248. J Clin Med. 2020. PMID: 32344923 Free PMC article.
References
-
- Ling Y, Johnson MK, Kiely DG, et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med 2012;186:790–796. - PubMed
-
- Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010;122:156–163. - PubMed
-
- Voelkel NF, Cool C. Pathology of pulmonary hypertension. Cardiol Clin 2004;22:343–351, v. - PubMed
-
- Davies RJ, Morrell NW. Molecular mechanisms of pulmonary arterial hypertension: role of mutations in the bone morphogenetic protein type II receptor. Chest 2008;134:1271–1277. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
