

Application of a Physiologically Based Pharmacokinetic Model to Predict OATP1B1-Related Variability in Pharmacodynamics of Rosuvastatin
- PMID: 25006781
- PMCID: PMC4120018
- DOI: 10.1038/psp.2014.24
Application of a Physiologically Based Pharmacokinetic Model to Predict OATP1B1-Related Variability in Pharmacodynamics of Rosuvastatin
Abstract
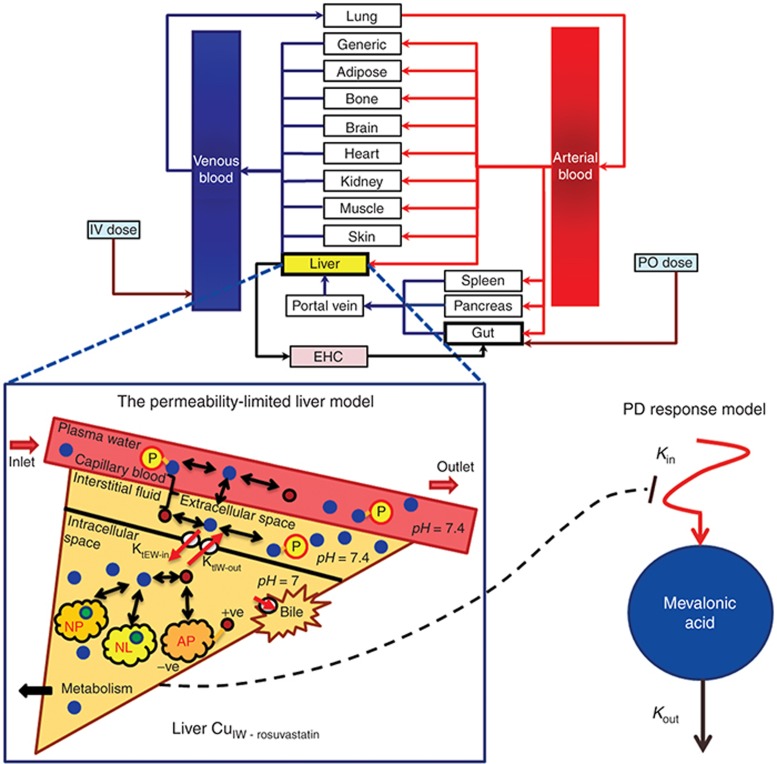
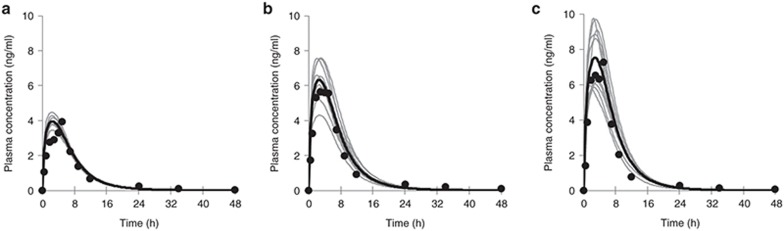
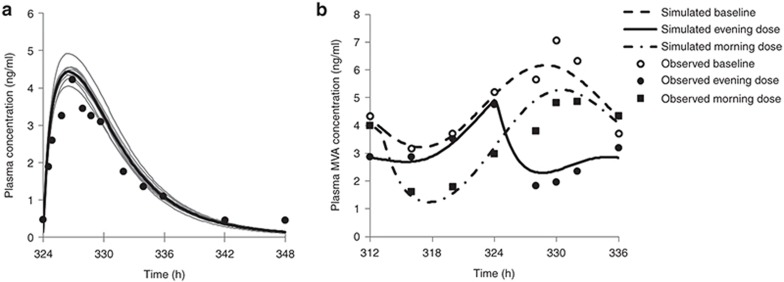
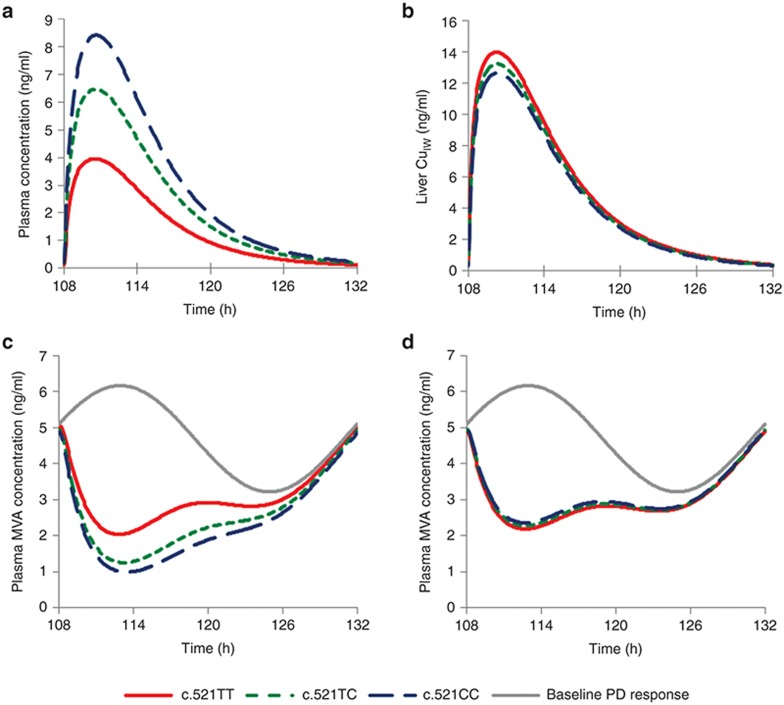
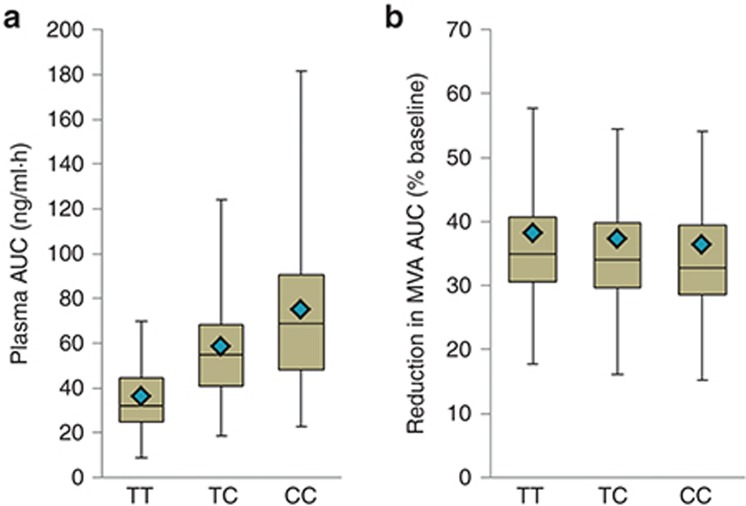
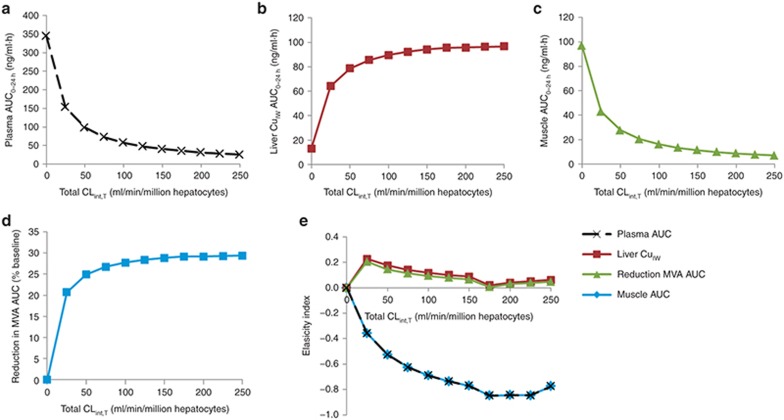
Typically, pharmacokinetic-pharmacodynamic (PK/PD) models use plasma concentration as the input that drives the PD model. However, interindividual variability in uptake transporter activity can lead to variable drug concentrations in plasma without discernible impact on the effect site organ concentration. A physiologically based PK/PD model for rosuvastatin was developed that linked the predicted liver concentration to the PD response model. The model was then applied to predict the effect of genotype-dependent uptake by the organic anion-transporting polypeptide 1B1 (OATP1B1) transporter on the pharmacological response. The area under the plasma concentration-time curve (AUC0-∞) was increased by 63 and 111% for the c.521TC and c.521CC genotypes vs. the c.521TT genotype, while the PD response remained relatively unchanged (3.1 and 5.8% reduction). Using local concentration at the effect site to drive the PD response enabled us to explain the observed disconnect between the effect of the OATP1B1 c521T>C polymorphism on rosuvastatin plasma concentration and the cholesterol synthesis response.
Figures
Similar articles
-
Effect of SLCO1B1 polymorphism on the plasma concentrations of bile acids and bile acid synthesis marker in humans.Pharmacogenet Genomics. 2009 Jun;19(6):447-57. doi: 10.1097/FPC.0b013e32832bcf7b. Pharmacogenet Genomics. 2009. PMID: 19387419
-
No significant effect of SLCO1B1 polymorphism on the pharmacokinetics of rosiglitazone and pioglitazone.Br J Clin Pharmacol. 2008 Jan;65(1):78-86. doi: 10.1111/j.1365-2125.2007.02986.x. Epub 2007 Jul 17. Br J Clin Pharmacol. 2008. PMID: 17635496 Free PMC article. Clinical Trial.
-
SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid.Pharmacogenet Genomics. 2006 Dec;16(12):873-9. doi: 10.1097/01.fpc.0000230416.82349.90. Pharmacogenet Genomics. 2006. PMID: 17108811
-
Genetic polymorphisms of uptake (OATP1B1, 1B3) and efflux (MRP2, BCRP) transporters: implications for inter-individual differences in the pharmacokinetics and pharmacodynamics of statins and other clinically relevant drugs.Expert Opin Drug Metab Toxicol. 2009 Jul;5(7):703-29. doi: 10.1517/17425250902976854. Expert Opin Drug Metab Toxicol. 2009. PMID: 19442037 Review.
-
SLCO1B1 polymorphism and oral antidiabetic drugs.Basic Clin Pharmacol Toxicol. 2010 Oct;107(4):775-81. doi: 10.1111/j.1742-7843.2010.00581.x. Basic Clin Pharmacol Toxicol. 2010. PMID: 20406215 Review.
Cited by
-
Top-down, Bottom-up and Middle-out Strategies for Drug Cardiac Safety Assessment via Modeling and Simulations.Curr Pharmacol Rep. 2016;2(4):171-177. doi: 10.1007/s40495-016-0060-3. Epub 2016 Apr 5. Curr Pharmacol Rep. 2016. PMID: 27429898 Free PMC article. Review.
-
Pharmacokinetic Considerations for Combining Antiretroviral Therapy, Direct-Acting Antiviral Agents for Hepatitis C Virus, and Addiction Treatment Medications.Clin Pharmacol Drug Dev. 2017 Mar;6(2):135-139. doi: 10.1002/cpdd.313. Clin Pharmacol Drug Dev. 2017. PMID: 28263465 Free PMC article. Review.
-
Advancing Predictions of Tissue and Intracellular Drug Concentrations Using In Vitro, Imaging and Physiologically Based Pharmacokinetic Modeling Approaches.Clin Pharmacol Ther. 2018 Nov;104(5):865-889. doi: 10.1002/cpt.1183. Epub 2018 Sep 12. Clin Pharmacol Ther. 2018. PMID: 30059145 Free PMC article.
-
Pharmacokinetics and Pharmacodynamics of Meloxicam in East Asian Populations: The Role of Ethnicity on Drug Response.CPT Pharmacometrics Syst Pharmacol. 2017 Dec;6(12):823-832. doi: 10.1002/psp4.12259. Epub 2017 Nov 23. CPT Pharmacometrics Syst Pharmacol. 2017. PMID: 29024493 Free PMC article.
-
Recent Advances in Development and Application of Physiologically-Based Pharmacokinetic (PBPK) Models: a Transition from Academic Curiosity to Regulatory Acceptance.Curr Pharmacol Rep. 2016;2(3):161-169. doi: 10.1007/s40495-016-0059-9. Epub 2016 Apr 14. Curr Pharmacol Rep. 2016. PMID: 27226953 Free PMC article. Review.
References
-
- Zhao P., Rowland M., Huang S.M. Best practice in the use of physiologically based pharmacokinetic modeling and simulation to address clinical pharmacology regulatory questions. Clin. Pharmacol. Ther. 2012;92:17–20. - PubMed
-
- Zhao P., et al. Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin. Pharmacol. Ther. 2011;89:259–267. - PubMed
-
- Perera V., Elmeliegy M.A., Rao G., Forrest A. The link between pharmacodynamics and physiologically based pharmacokinetic models. Clin. Pharmacol. Ther. 2013;93:151–152. - PubMed
-
- Rostami-Hodjegan A. Response to “The link between pharmacodynamics and physiologically based pharmacokinetic models.”. Clin. Pharmacol. Ther. 2013;93:152. - PubMed
-
- Watanabe T., Kusuhara H., Maeda K., Shitara Y., Sugiyama Y. Physiologically based pharmacokinetic modeling to predict transporter-mediated clearance and distribution of pravastatin in humans. J. Pharmacol. Exp. Ther. 2009;328:652–662. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
