

Osteogenesis imperfecta due to mutations in non-collagenous genes: lessons in the biology of bone formation
- PMID: 25007323
- PMCID: PMC4183132
- DOI: 10.1097/MOP.0000000000000117
Osteogenesis imperfecta due to mutations in non-collagenous genes: lessons in the biology of bone formation
Abstract
Purpose of review: Osteogenesis imperfecta or 'brittle bone disease' has mainly been considered a bone disorder caused by collagen mutations. Within the last decade, however, a surge of genetic discoveries has created a new paradigm for osteogenesis imperfecta as a collagen-related disorder, where most cases are due to autosomal dominant type I collagen defects, while rare, mostly recessive, forms are due to defects in genes whose protein products interact with collagen protein. This review is both timely and relevant in outlining the genesis, development, and future of this paradigm shift in the understanding of osteogenesis imperfecta.
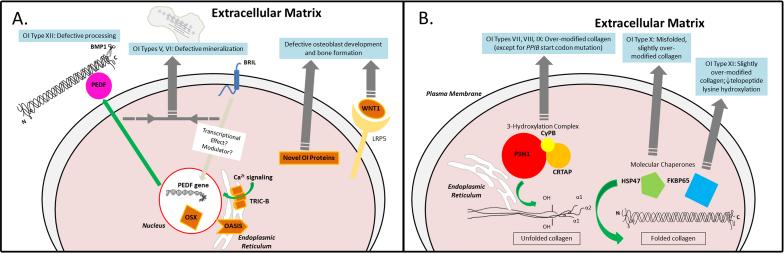
Recent findings: Bone-restricted interferon-induced transmembrane (IFITM)-like protein (BRIL) and pigment epithelium-derived factor (PEDF) defects cause types V and VI osteogenesis imperfecta via defective bone mineralization, while defects in cartilage-associated protein (CRTAP), prolyl 3-hydroxylase 1 (P3H1), and cyclophilin B (CYPB) cause types VII-IX osteogenesis imperfecta via defective collagen post-translational modification. Heat shock protein 47 (HSP47) and FK506-binding protein-65 (FKBP65) defects cause types X and XI osteogenesis imperfecta via aberrant collagen crosslinking, folding, and chaperoning, while defects in SP7 transcription factor, wingless-type MMTV integration site family member 1 (WNT1), trimeric intracellular cation channel type b (TRIC-B), and old astrocyte specifically induced substance (OASIS) disrupt osteoblast development. Finally, absence of the type I collagen C-propeptidase bone morphogenetic protein 1 (BMP1) causes type XII osteogenesis imperfecta due to altered collagen maturation/processing.
Summary: Identification of these multiple causative defects has provided crucial information for accurate genetic counseling, inspired a recently proposed functional grouping of osteogenesis imperfecta types by shared mechanism to simplify current nosology, and has prodded investigations into common pathways in osteogenesis imperfecta. Such investigations could yield critical information on cellular and bone tissue mechanisms and translate to new mechanistic insight into clinical therapies for patients.
Figures
References
-
- Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S, et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat. 2007 Mar;28(3):209–21. - PMC - PubMed
-
- Glorieux FH, Moffatt P. Osteogenesis imperfecta, an ever-expanding conundrum. J Bone Miner Res. 2013 Jul;28(7):1519–22. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
