

Abnormalities in human pluripotent cells due to reprogramming mechanisms
- PMID: 25008523
- PMCID: PMC4898064
- DOI: 10.1038/nature13551
Abnormalities in human pluripotent cells due to reprogramming mechanisms
Abstract
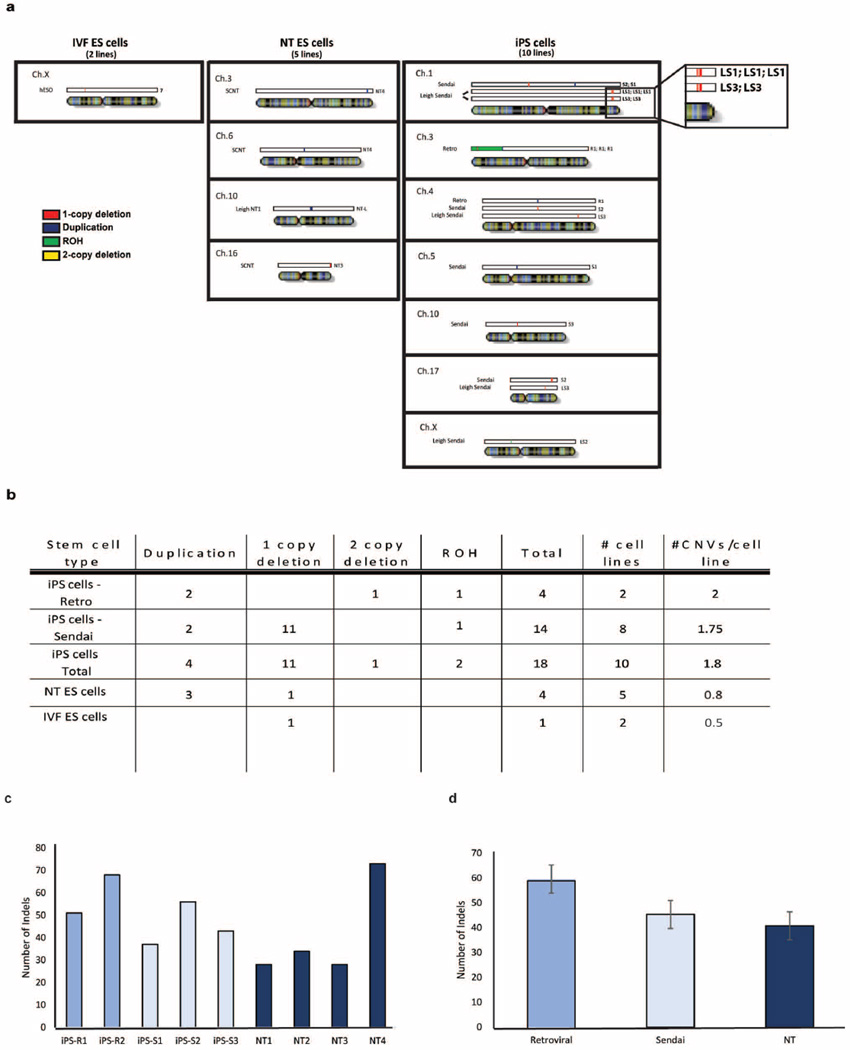
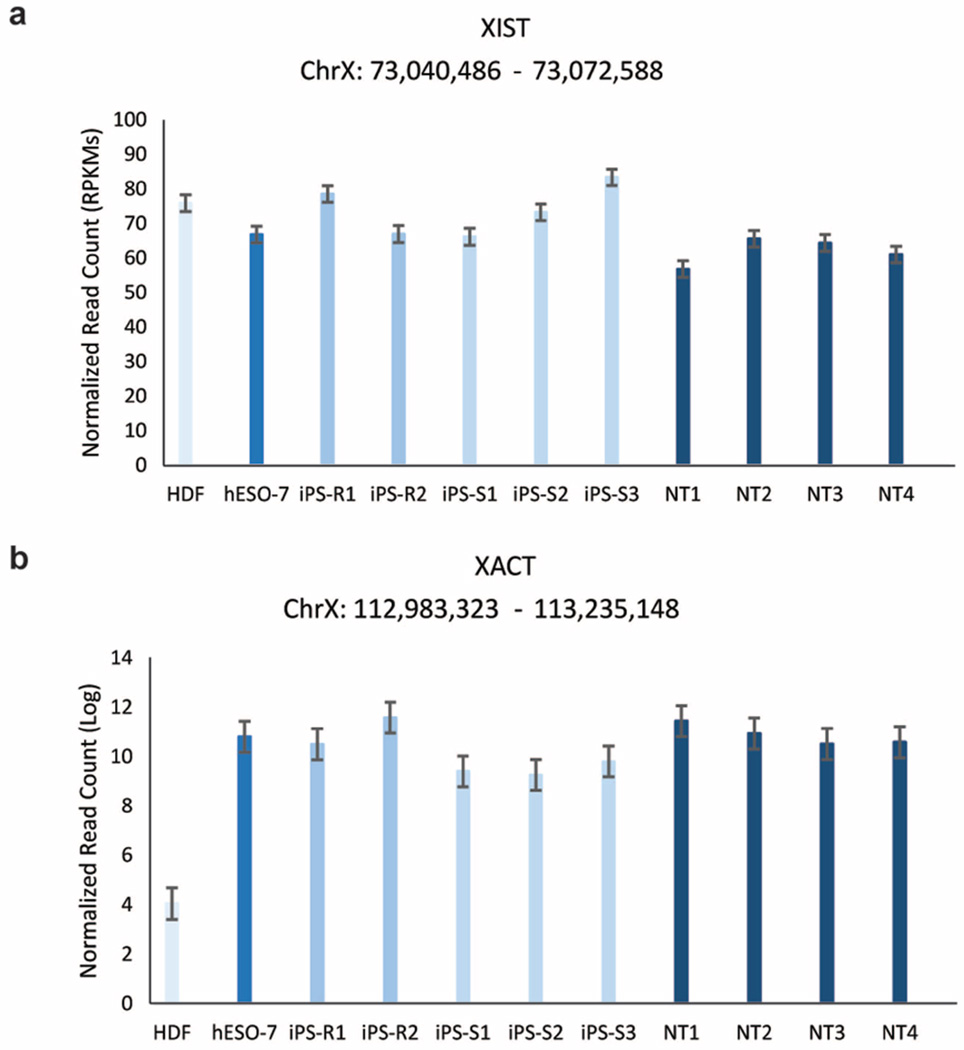
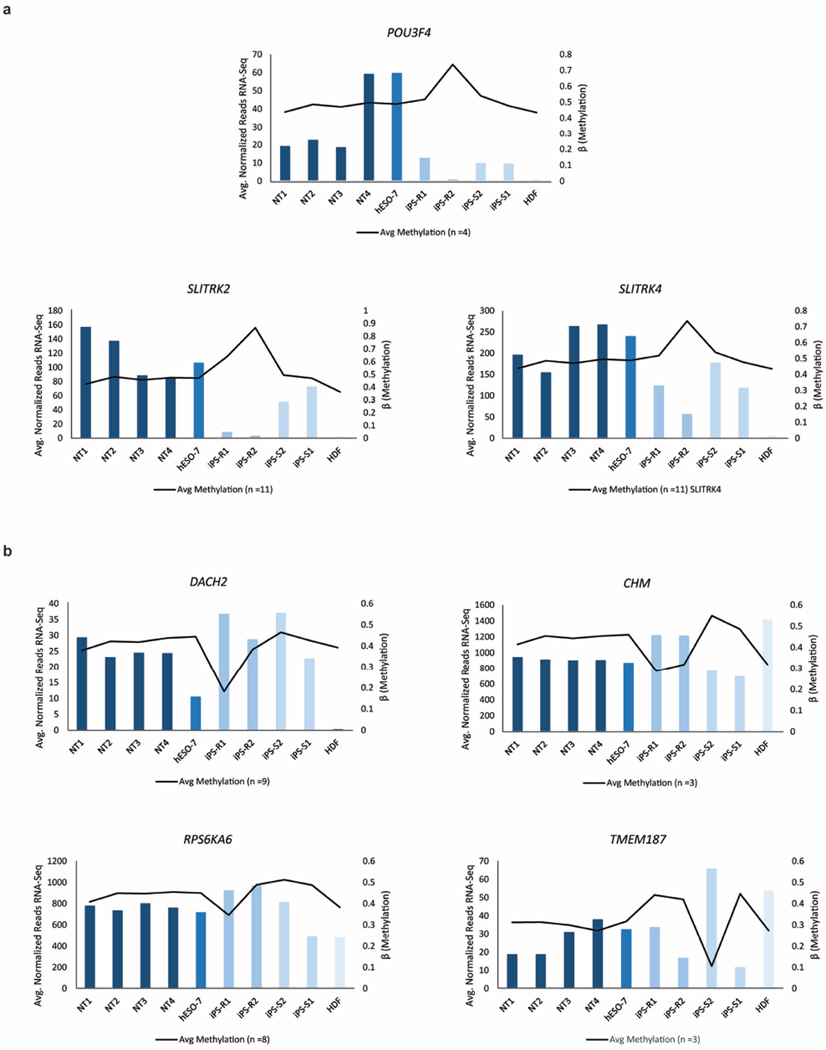
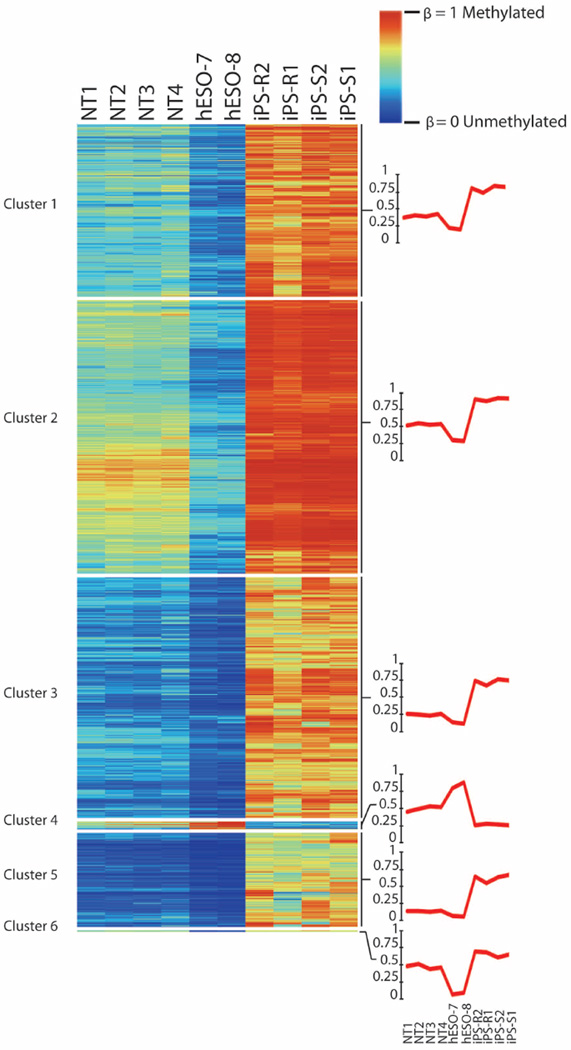
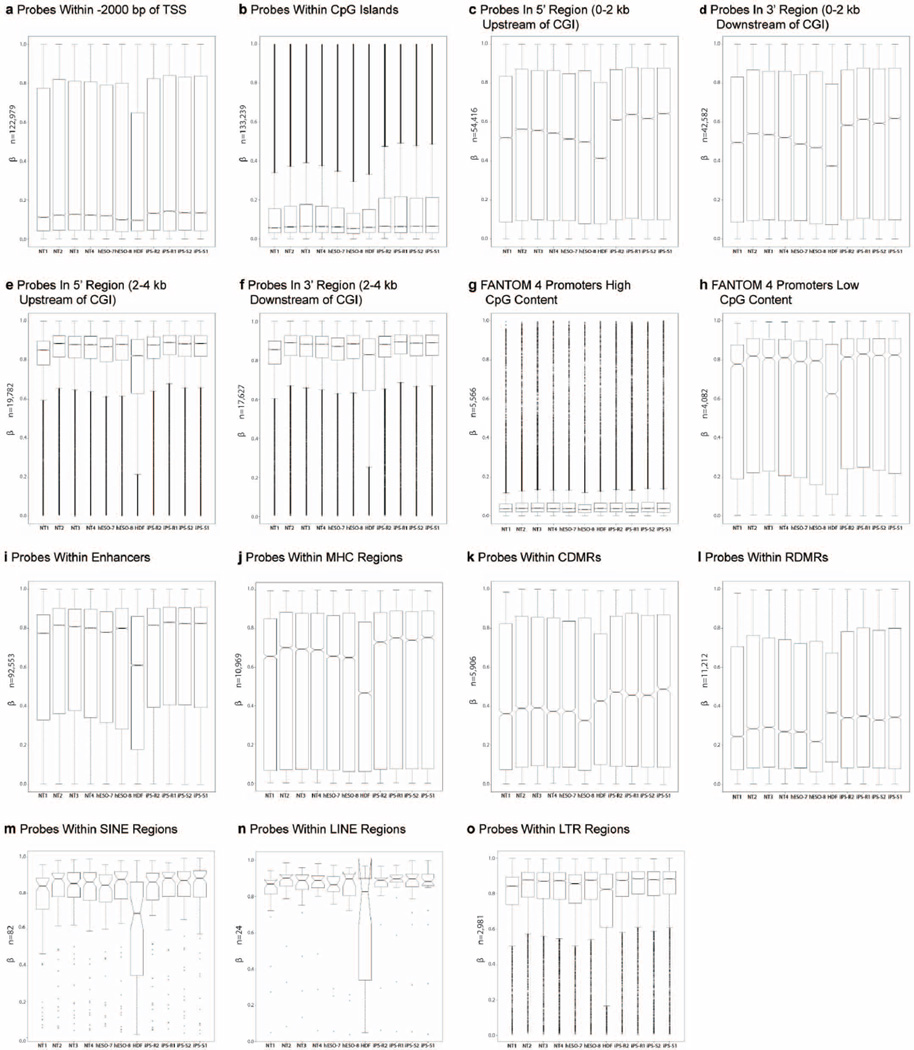
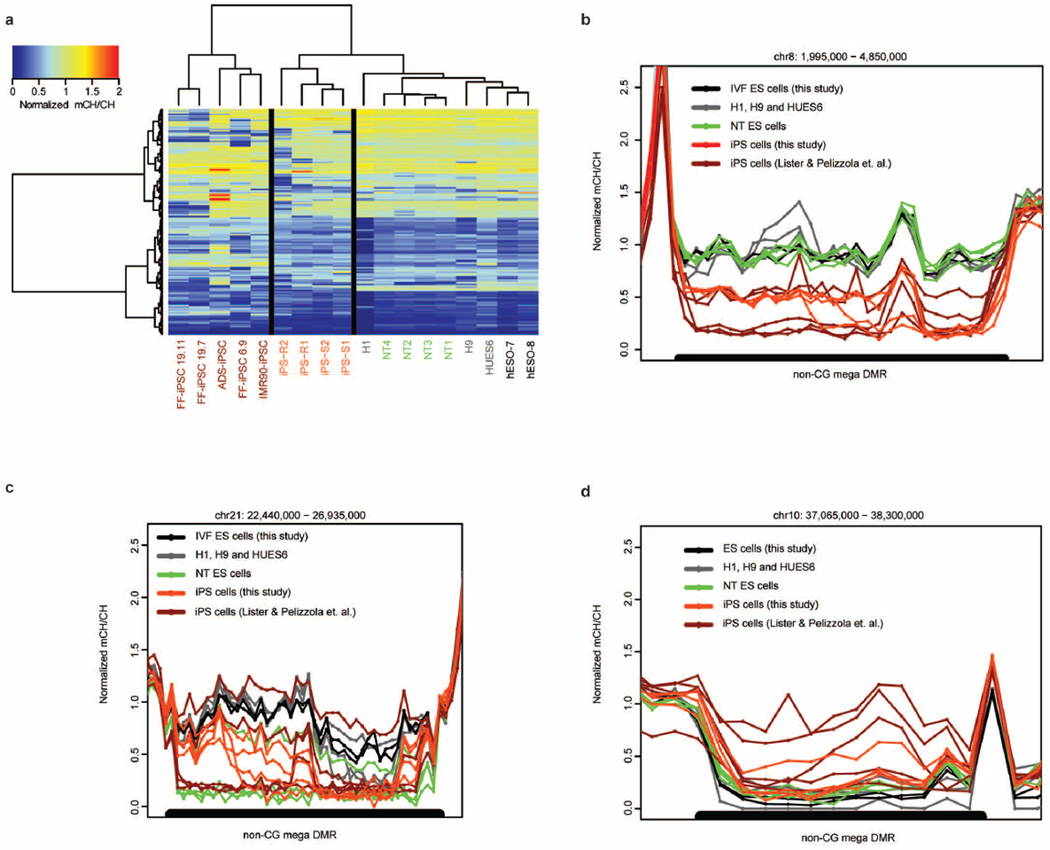
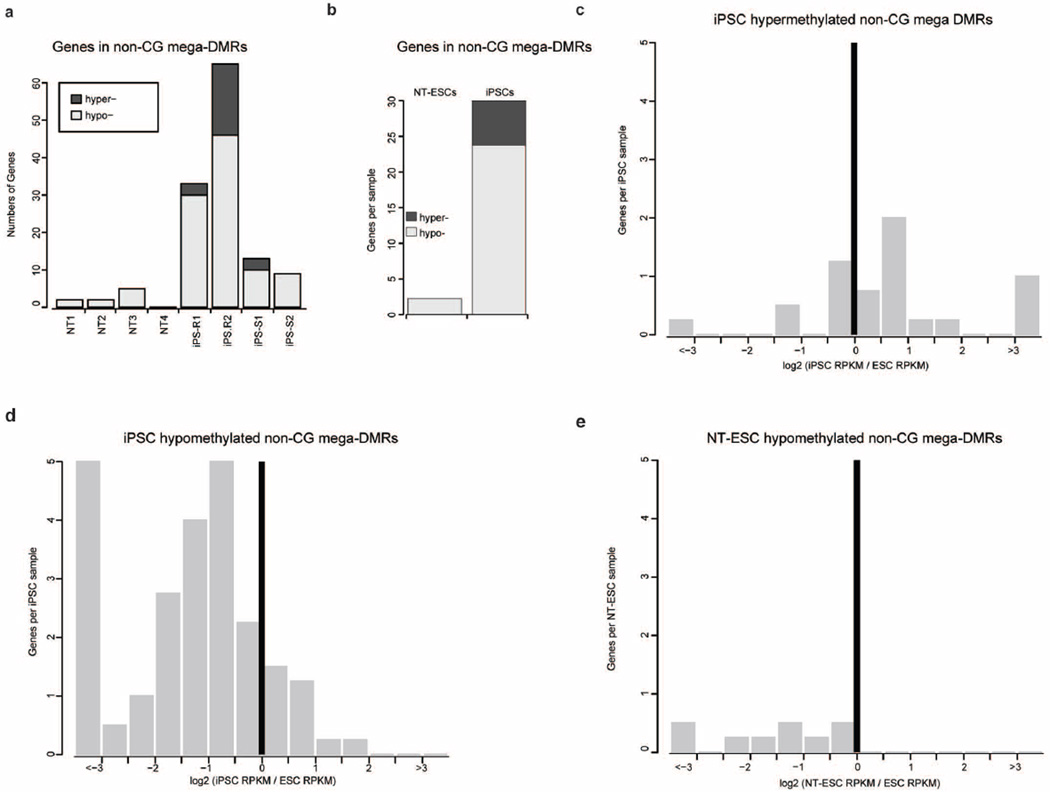
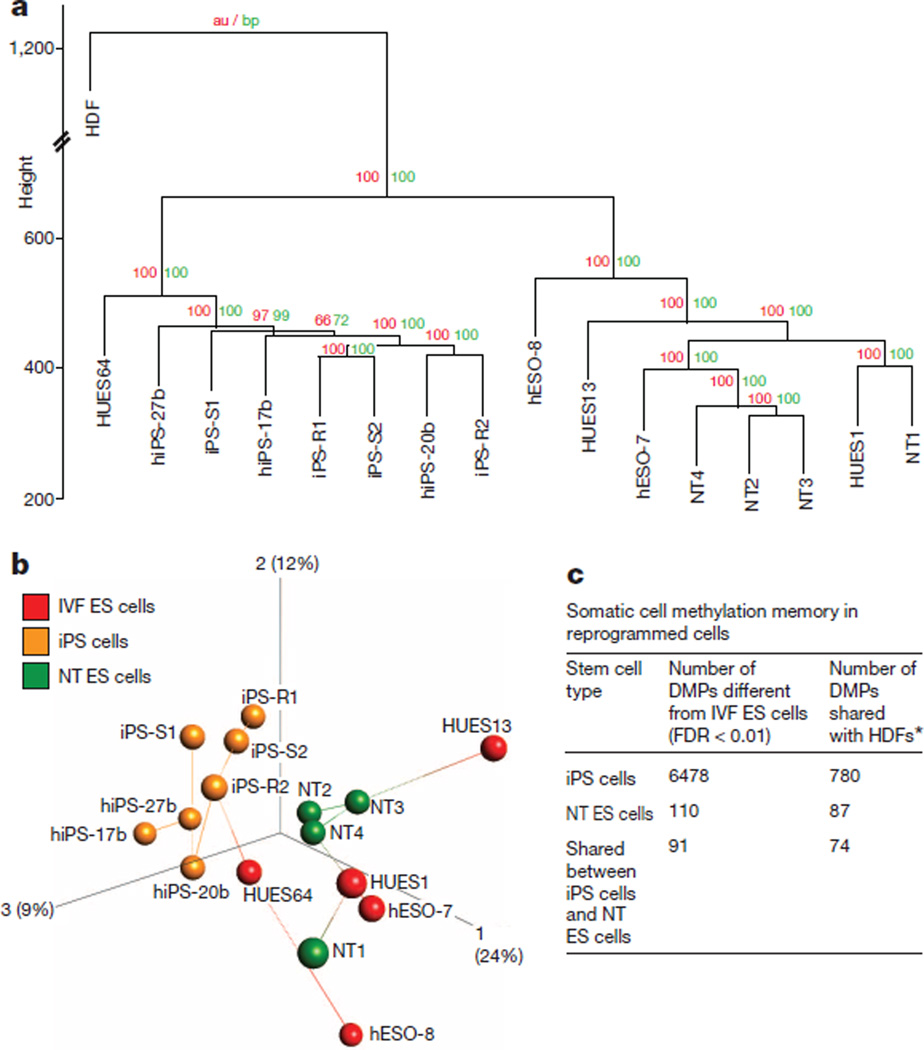
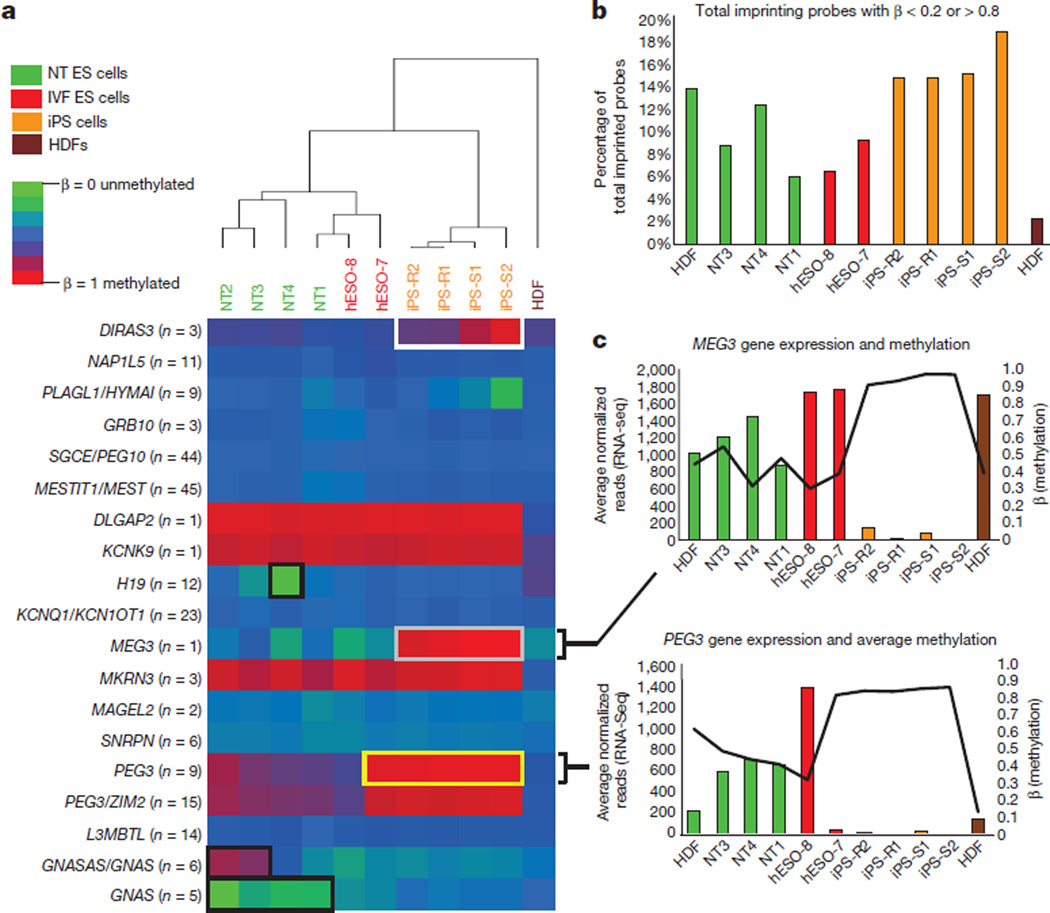
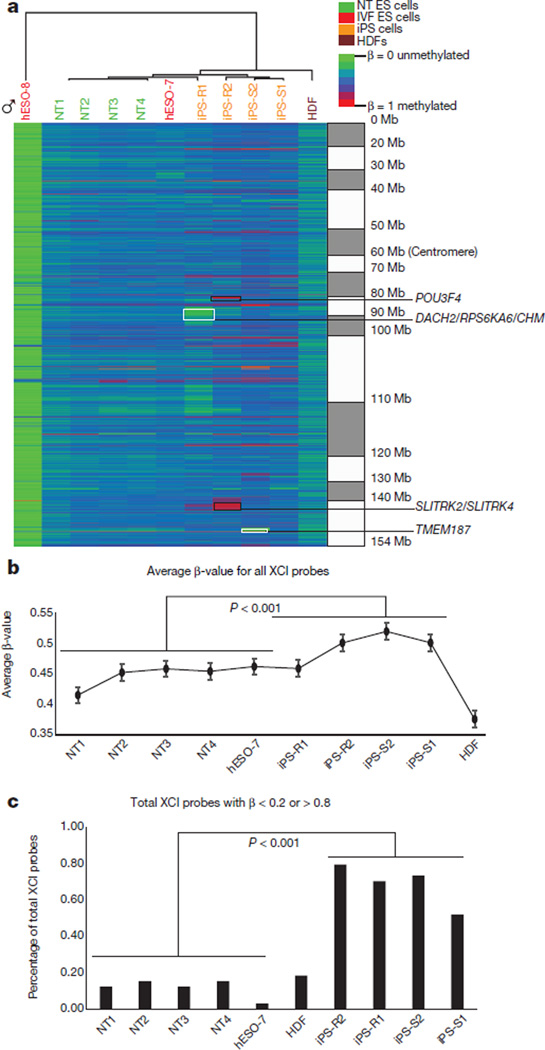
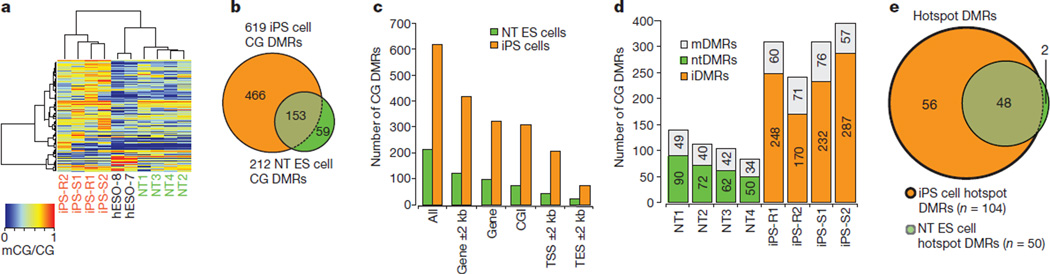
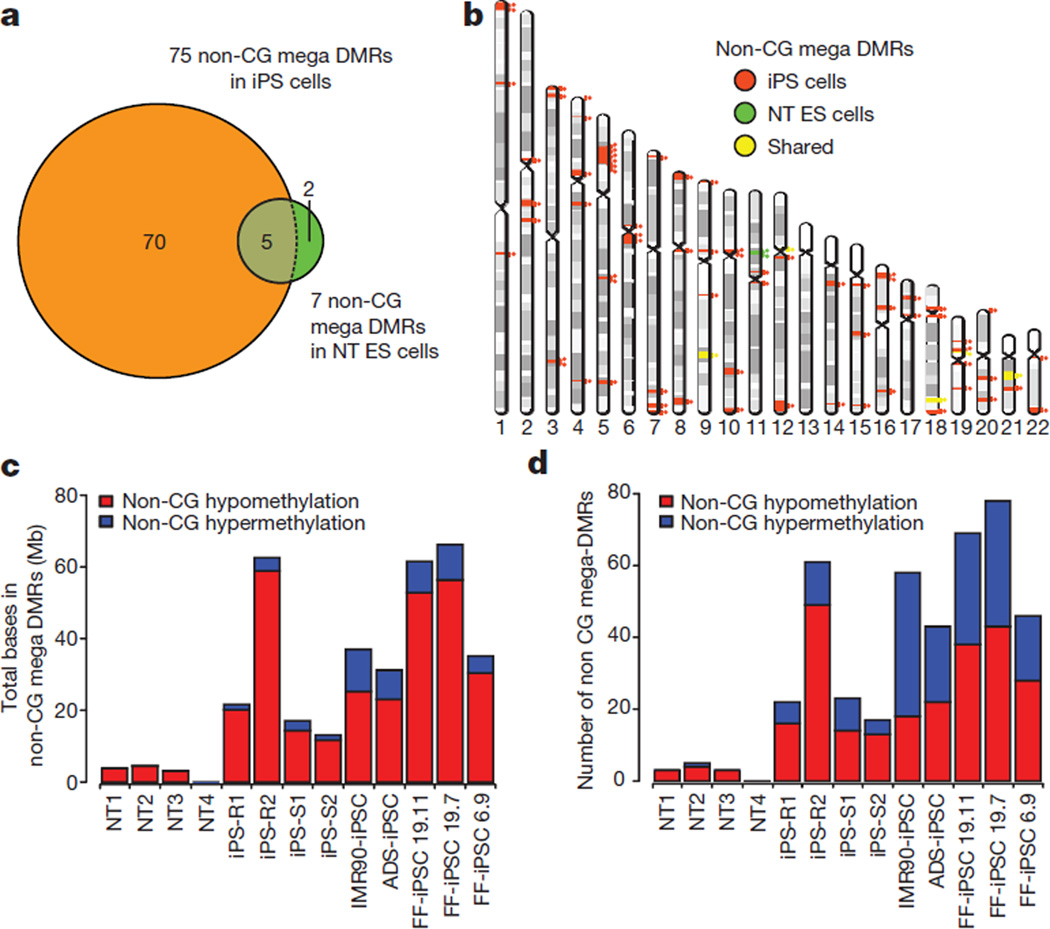
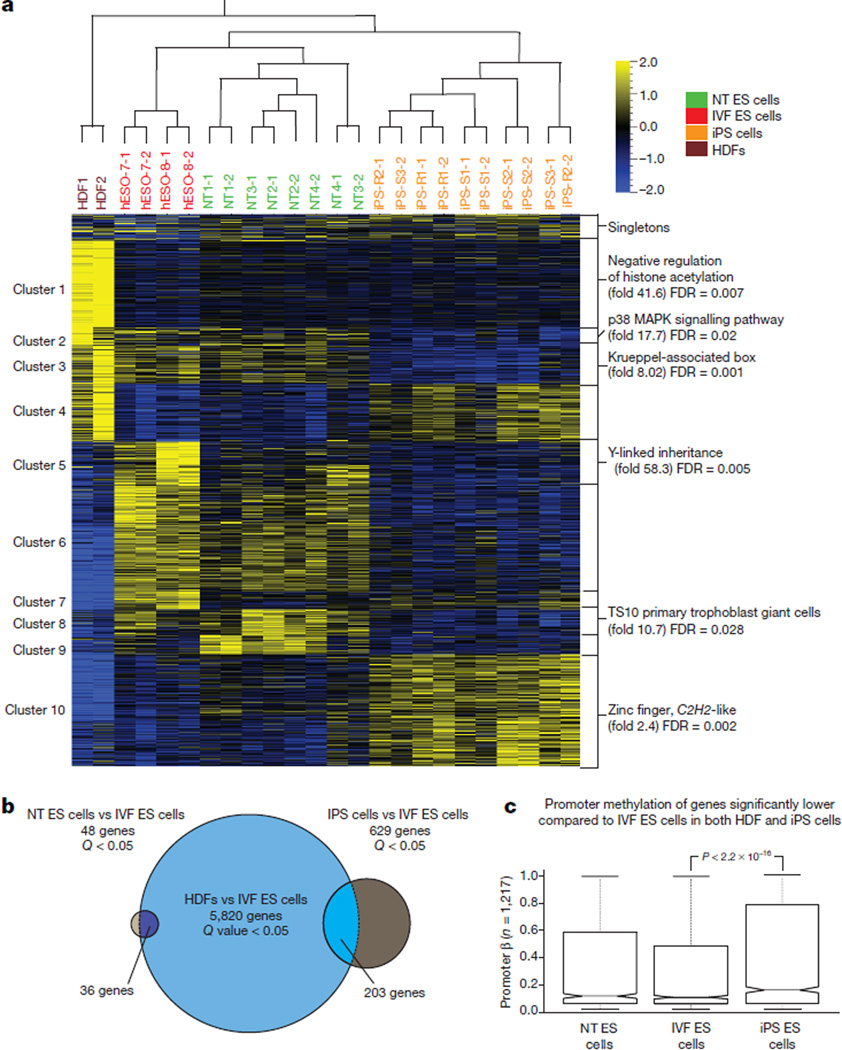
Human pluripotent stem cells hold potential for regenerative medicine, but available cell types have significant limitations. Although embryonic stem cells (ES cells) from in vitro fertilized embryos (IVF ES cells) represent the 'gold standard', they are allogeneic to patients. Autologous induced pluripotent stem cells (iPS cells) are prone to epigenetic and transcriptional aberrations. To determine whether such abnormalities are intrinsic to somatic cell reprogramming or secondary to the reprogramming method, genetically matched sets of human IVF ES cells, iPS cells and nuclear transfer ES cells (NT ES cells) derived by somatic cell nuclear transfer (SCNT) were subjected to genome-wide analyses. Both NT ES cells and iPS cells derived from the same somatic cells contained comparable numbers of de novo copy number variations. In contrast, DNA methylation and transcriptome profiles of NT ES cells corresponded closely to those of IVF ES cells, whereas iPS cells differed and retained residual DNA methylation patterns typical of parental somatic cells. Thus, human somatic cells can be faithfully reprogrammed to pluripotency by SCNT and are therefore ideal for cell replacement therapies.
Figures

Comment in
-
Stem cells: The quest for the perfect reprogrammed cell.Nature. 2014 Jul 10;511(7508):160-2. doi: 10.1038/nature13515. Epub 2014 Jul 2. Nature. 2014. PMID: 25008516 No abstract available.
References
-
- Thomson JA, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. - PubMed
-
- Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. - PubMed
-
- Rais Y, et al. Deterministic direct reprogramming of somatic cells to pluripotency. Nature. 2013;502:65–70. - PubMed
-
- Hussein SM, et al. Copy number variation and selection during reprogramming to pluripotency. Nature. 2011;471:58–62. - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
