

Different design of enzyme-triggered CO-releasing molecules (ET-CORMs) reveals quantitative differences in biological activities in terms of toxicity and inflammation
- PMID: 25009775
- PMCID: PMC4085349
- DOI: 10.1016/j.redox.2014.06.002
Different design of enzyme-triggered CO-releasing molecules (ET-CORMs) reveals quantitative differences in biological activities in terms of toxicity and inflammation
Abstract
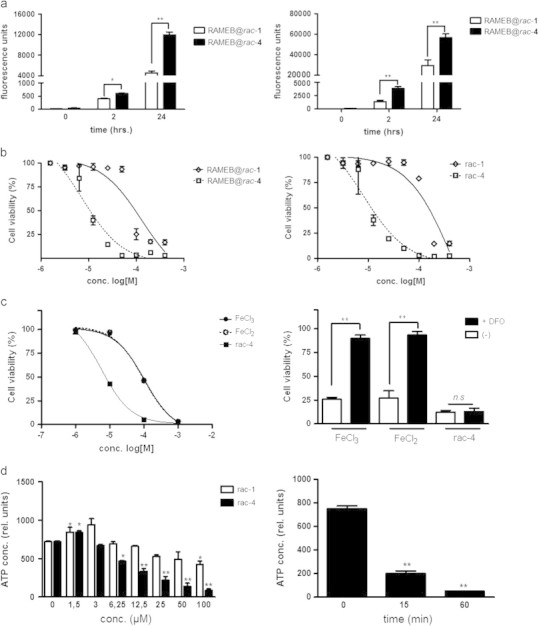
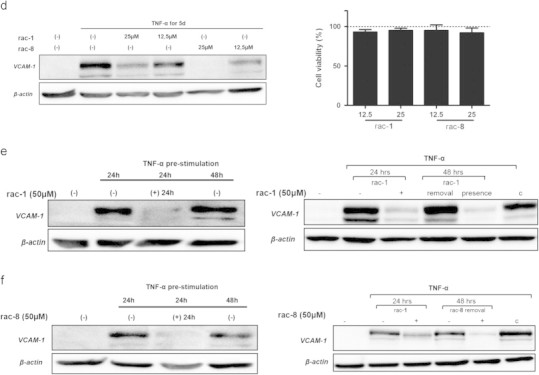
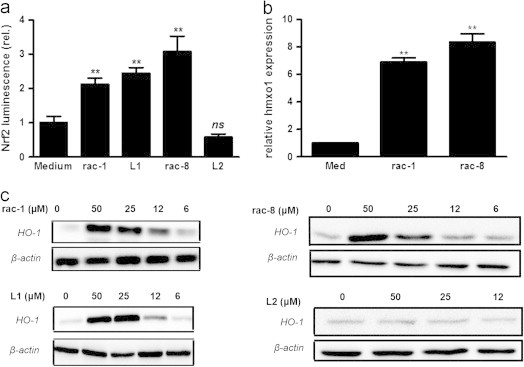
Acyloxydiene-Fe(CO)3 complexes can act as enzyme-triggered CO-releasing molecules (ET-CORMs). Their biological activity strongly depends on the mother compound from which they are derived, i.e. cyclohexenone or cyclohexanedione, and on the position of the ester functionality they harbour. The present study addresses if the latter characteristic affects CO release, if cytotoxicity of ET-CORMs is mediated through iron release or inhibition of cell respiration and to what extent cyclohexenone and cyclohexanedione derived ET-CORMs differ in their ability to counteract TNF-α mediated inflammation. Irrespective of the formulation (DMSO or cyclodextrin), toxicity in HUVEC was significantly higher for ET-CORMs bearing the ester functionality at the outer (rac-4), as compared to the inner (rac-1) position of the cyclohexenone moiety. This was paralleled by an increased CO release from the former ET-CORM. Toxicity was not mediated via iron as EC50 values for rac-4 were significantly lower than for FeCl2 or FeCl3 and were not influenced by iron chelation. ATP depletion preceded toxicity suggesting impaired cell respiration as putative cause for cell death. In long-term HUVEC cultures inhibition of VCAM-1 expression by rac-1 waned in time, while for the cyclohexanedione derived rac-8 inhibition seems to increase. NFκB was inhibited by both rac-1 and rac-8 independent of IκBα degradation. Both ET-CORMs activated Nrf-2 and consequently induced the expression of HO-1. This study further provides a rational framework for designing acyloxydiene-Fe(CO)3 complexes as ET-CORMs with differential CO release and biological activities. We also provide a better understanding of how these complexes affect cell-biology in mechanistic terms.
Keywords: Adhesion molecules; CO, carbon monoxide; Carbon monoxide; ET-CORM, enzyme-triggered carbon monoxide-releasing molecule; Endothelial cells; Enzyme-triggered CORMs; HO-1, haem oxygenase 1; HUVEC, human umbilical vein endothelial cells; NFκΒ, nuclear factor kappa-light-chain enhancer of activated B-cells; Nrf2, nuclear factor(erythroid-derived); TNF-α, tumour necrosis factor alpha; VCAM-1, vascular cell adhesion molecule 1.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
