

The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation
- PMID: 25010205
- PMCID: PMC4091782
- DOI: 10.1371/journal.pgen.1004475
The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation
Erratum in
- PLoS Genet. 2014 Aug;10(8):e1004629
Abstract
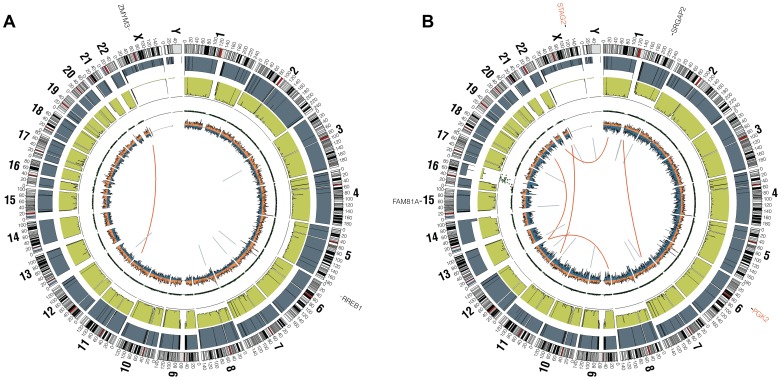
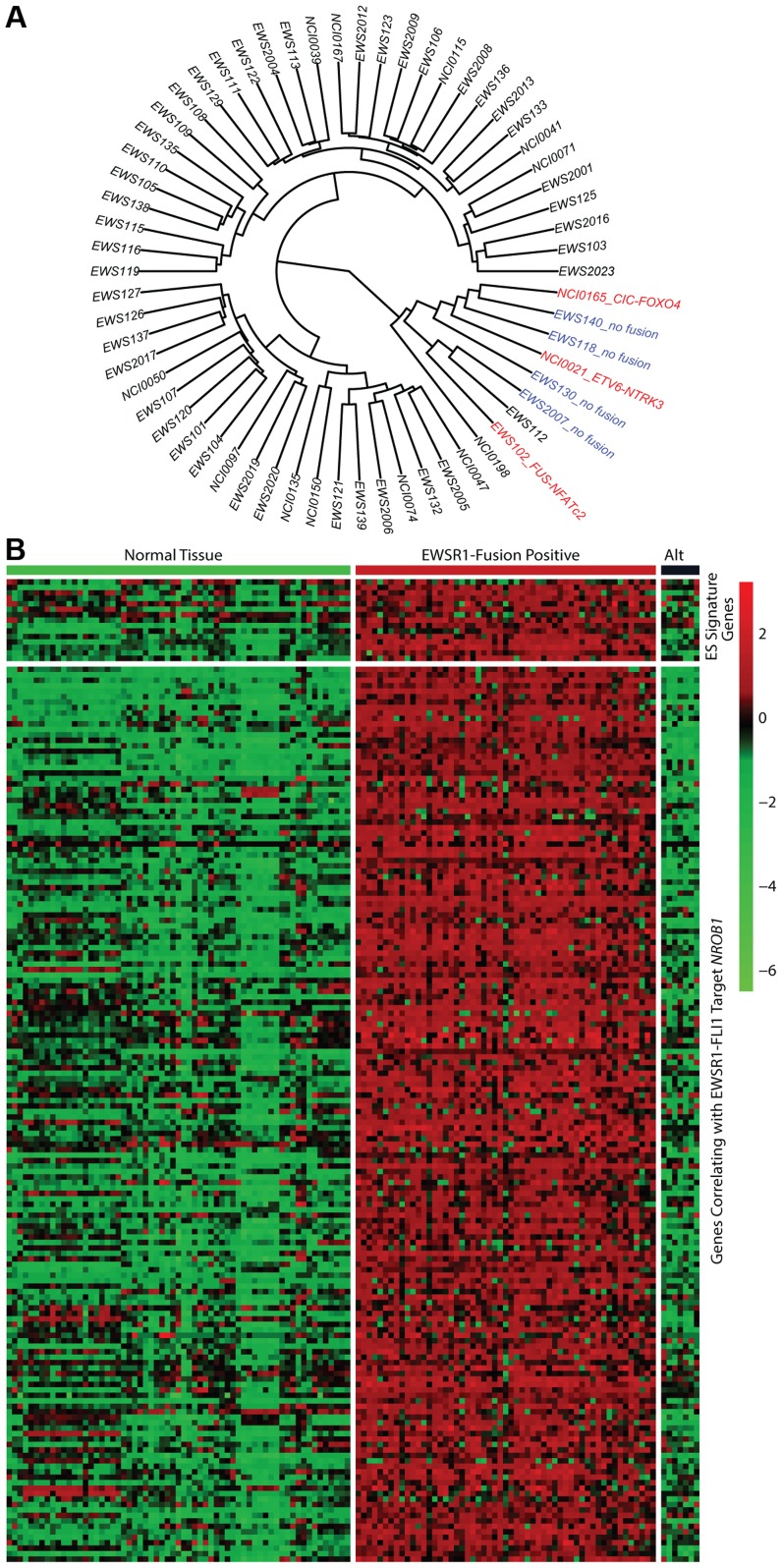
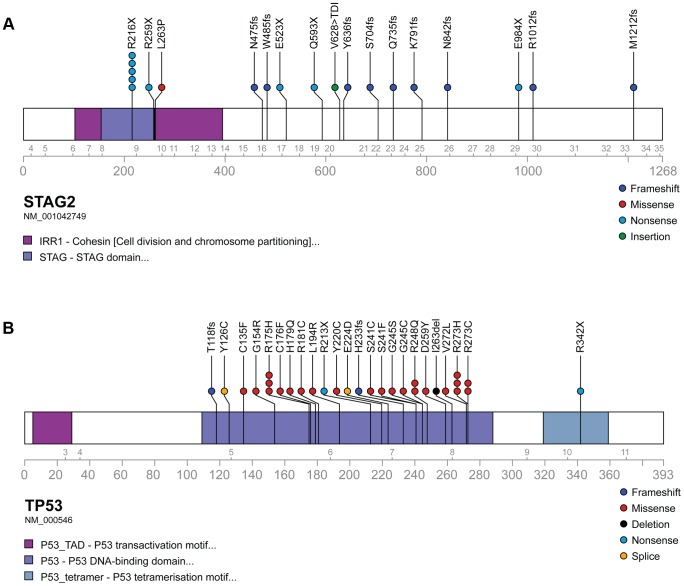
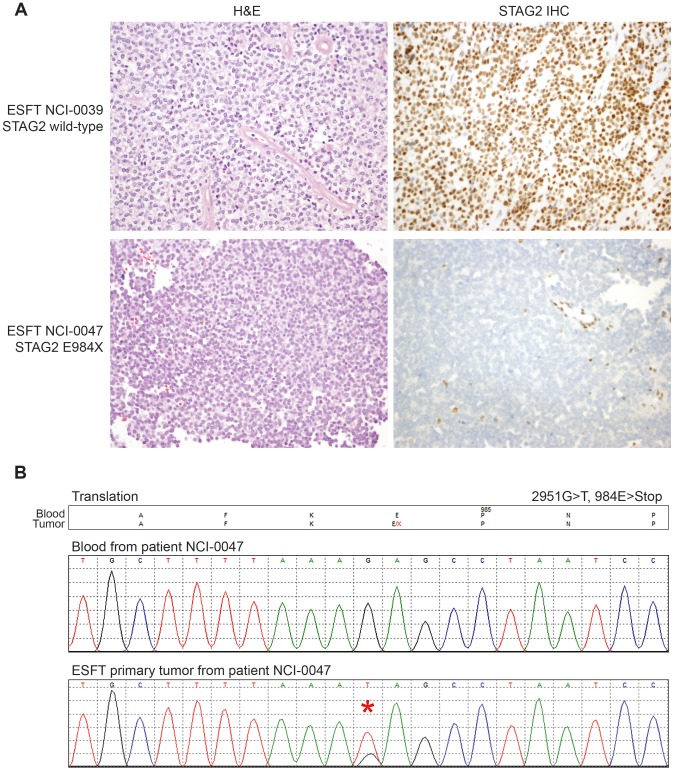
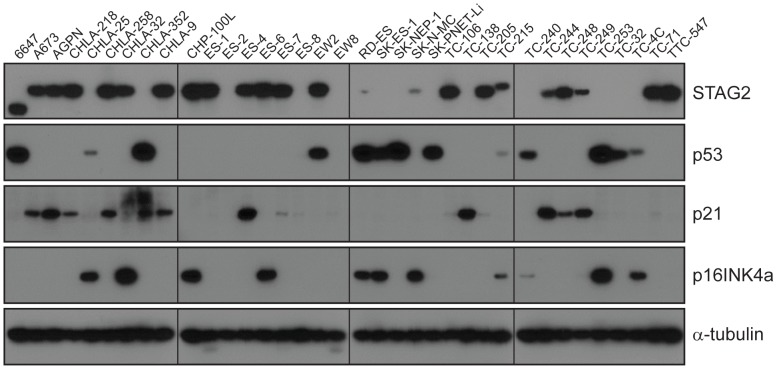
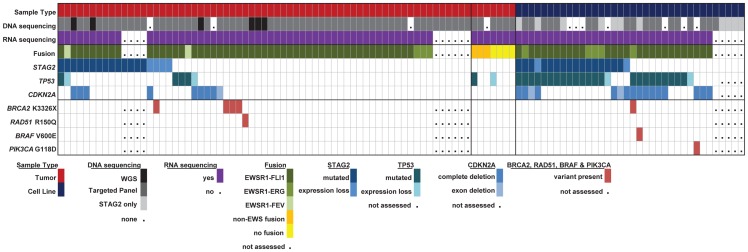
The Ewing sarcoma family of tumors (EFT) is a group of highly malignant small round blue cell tumors occurring in children and young adults. We report here the largest genomic survey to date of 101 EFT (65 tumors and 36 cell lines). Using a combination of whole genome sequencing and targeted sequencing approaches, we discover that EFT has a very low mutational burden (0.15 mutations/Mb) but frequent deleterious mutations in the cohesin complex subunit STAG2 (21.5% tumors, 44.4% cell lines), homozygous deletion of CDKN2A (13.8% and 50%) and mutations of TP53 (6.2% and 71.9%). We additionally note an increased prevalence of the BRCA2 K3326X polymorphism in EFT patient samples (7.3%) compared to population data (OR 7.1, p = 0.006). Using whole transcriptome sequencing, we find that 11% of tumors pathologically diagnosed as EFT lack a typical EWSR1 fusion oncogene and that these tumors do not have a characteristic Ewing sarcoma gene expression signature. We identify samples harboring novel fusion genes including FUS-NCATc2 and CIC-FOXO4 that may represent distinct small round blue cell tumor variants. In an independent EFT tissue microarray cohort, we show that STAG2 loss as detected by immunohistochemistry may be associated with more advanced disease (p = 0.15) and a modest decrease in overall survival (p = 0.10). These results significantly advance our understanding of the genomic and molecular underpinnings of Ewing sarcoma and provide a foundation towards further efforts to improve diagnosis, prognosis, and precision therapeutics testing.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Howlader N, Noone A, Krapcho M, Garshell J, Neyman N, et al. (2013) SEER Cancer Statistics Review (1975–2010). Available: http://seer.cancer.gov/csr/1975_2010/. Accessed 11 June 2014.
-
- Barker LM, Pendergrass TW, Sanders JE, Hawkins DS (2005) Survival after recurrence of Ewing's sarcoma family of tumors. J Clin Oncol 23: 4354–4362. - PubMed
-
- Esiashvili N, Goodman M, Marcus RB Jr (2008) Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J Pediatr Hematol Oncol 30: 425–430. - PubMed
-
- Delattre O, Zucman J, Melot T, Garau XS, Zucker JM, et al. (1994) The Ewing family of tumors–a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 331: 294–299. - PubMed
-
- Turc-Carel C, Aurias A, Mugneret F, Lizard S, Sidaner I, et al. (1988) Chromosomes in Ewing's sarcoma. I. An evaluation of 85 cases of remarkable consistency of t(11;22)(q24;q12). Cancer Genet Cytogenet 32: 229–238. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
