

Clonal architecture of secondary acute myeloid leukemia defined by single-cell sequencing
- PMID: 25010716
- PMCID: PMC4091781
- DOI: 10.1371/journal.pgen.1004462
Clonal architecture of secondary acute myeloid leukemia defined by single-cell sequencing
Abstract
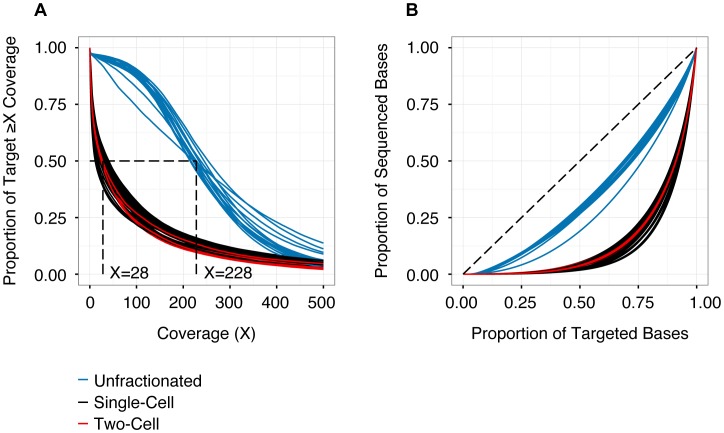
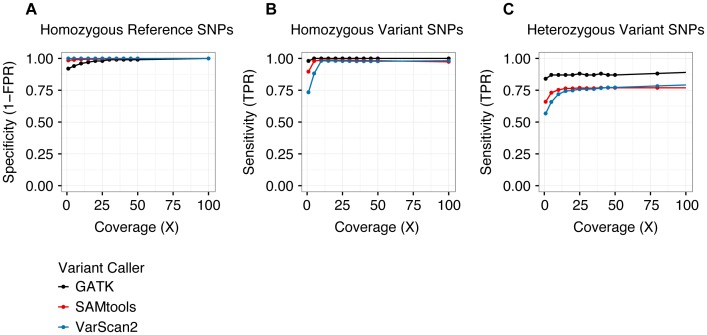
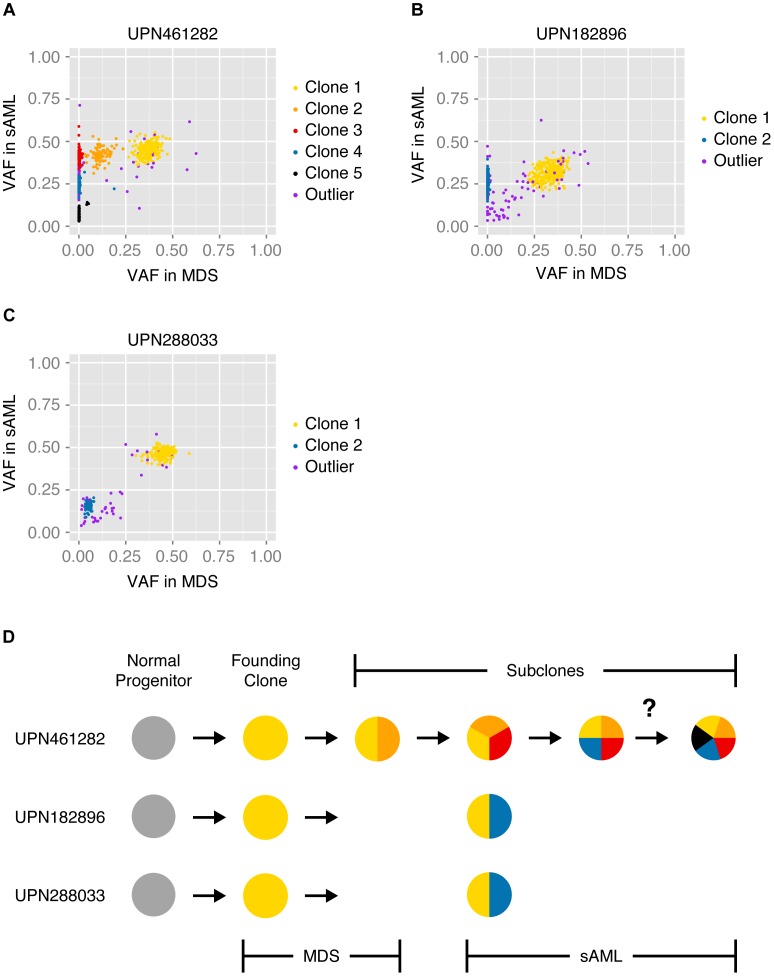
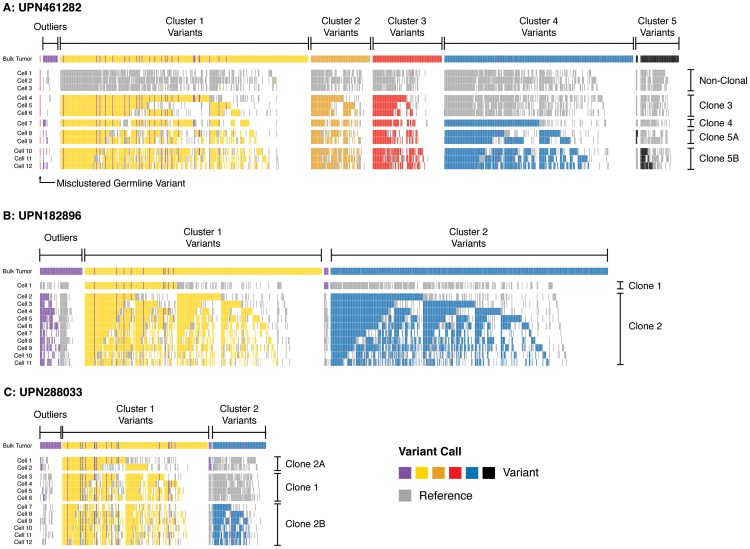
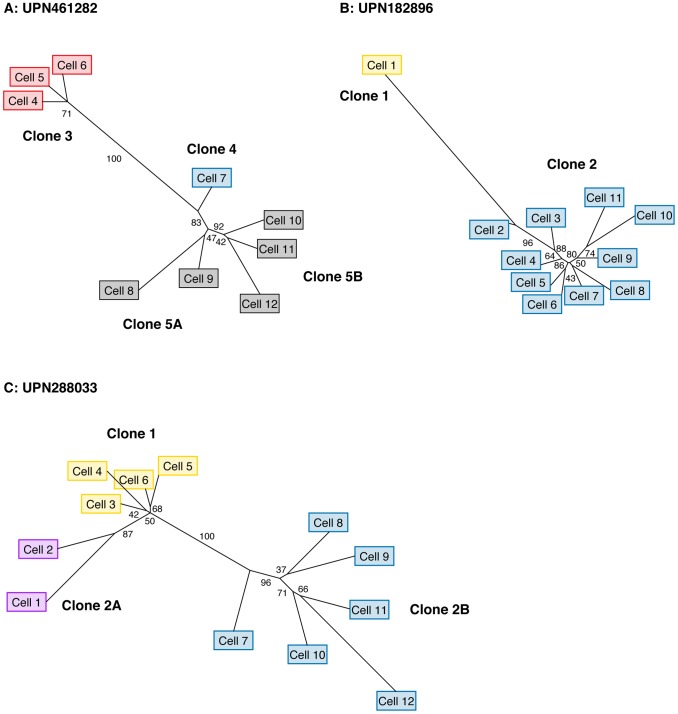
Next-generation sequencing has been used to infer the clonality of heterogeneous tumor samples. These analyses yield specific predictions-the population frequency of individual clones, their genetic composition, and their evolutionary relationships-which we set out to test by sequencing individual cells from three subjects diagnosed with secondary acute myeloid leukemia, each of whom had been previously characterized by whole genome sequencing of unfractionated tumor samples. Single-cell mutation profiling strongly supported the clonal architecture implied by the analysis of bulk material. In addition, it resolved the clonal assignment of single nucleotide variants that had been initially ambiguous and identified areas of previously unappreciated complexity. Accordingly, we find that many of the key assumptions underlying the analysis of tumor clonality by deep sequencing of unfractionated material are valid. Furthermore, we illustrate a single-cell sequencing strategy for interrogating the clonal relationships among known variants that is cost-effective, scalable, and adaptable to the analysis of both hematopoietic and solid tumors, or any heterogeneous population of cells.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
