

Renal angiotensin-converting enzyme is essential for the hypertension induced by nitric oxide synthesis inhibition
- PMID: 25012170
- PMCID: PMC4243348
- DOI: 10.1681/ASN.2013091030
Renal angiotensin-converting enzyme is essential for the hypertension induced by nitric oxide synthesis inhibition
Abstract
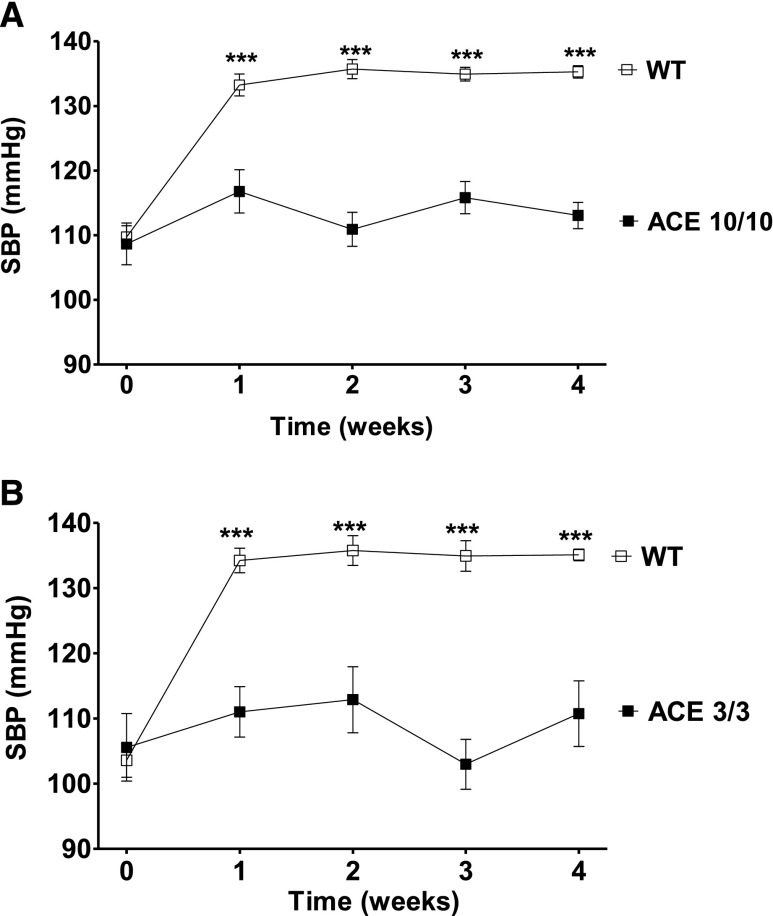
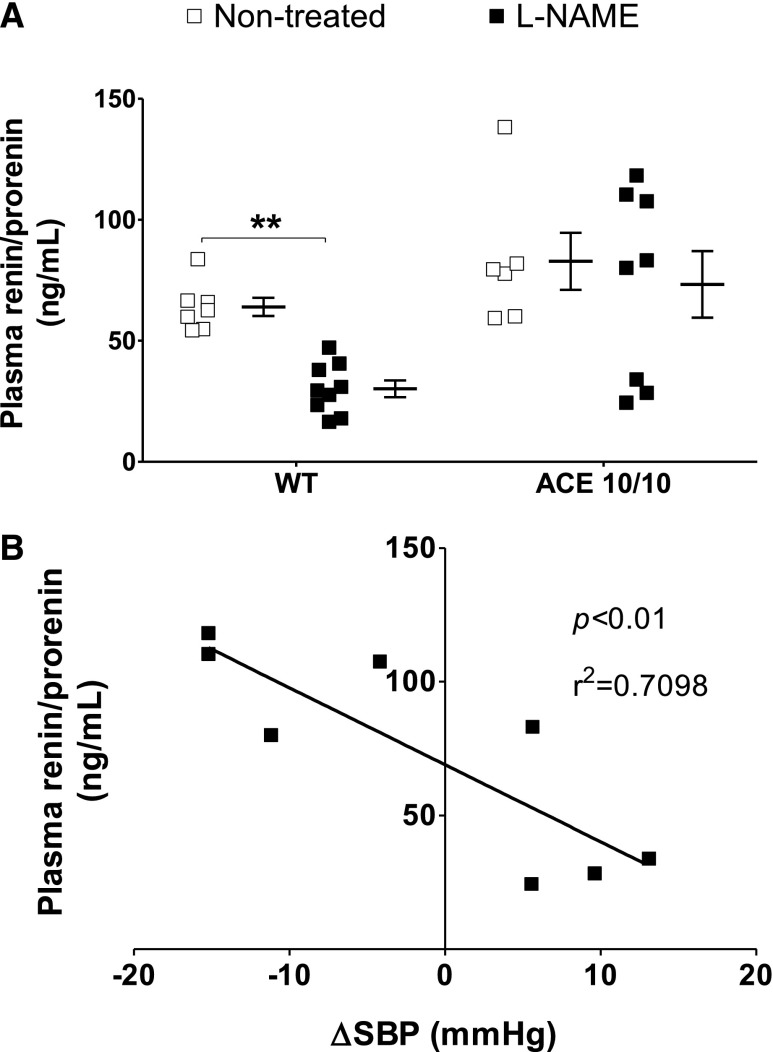
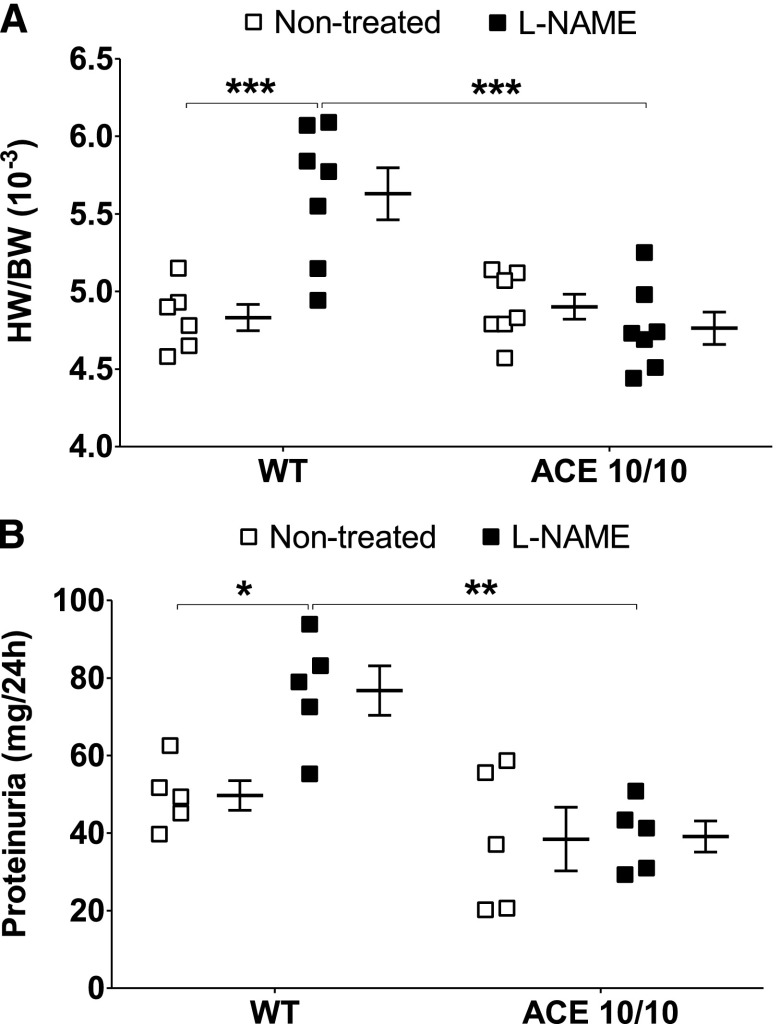
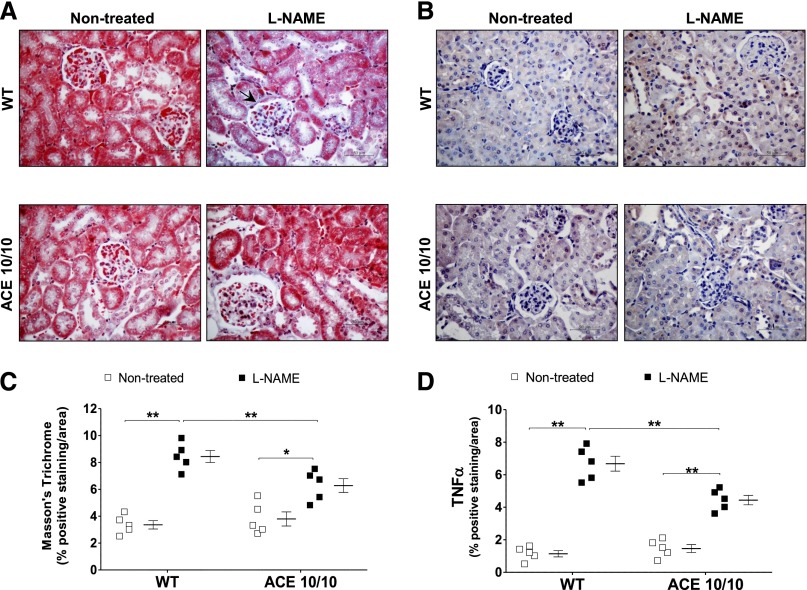
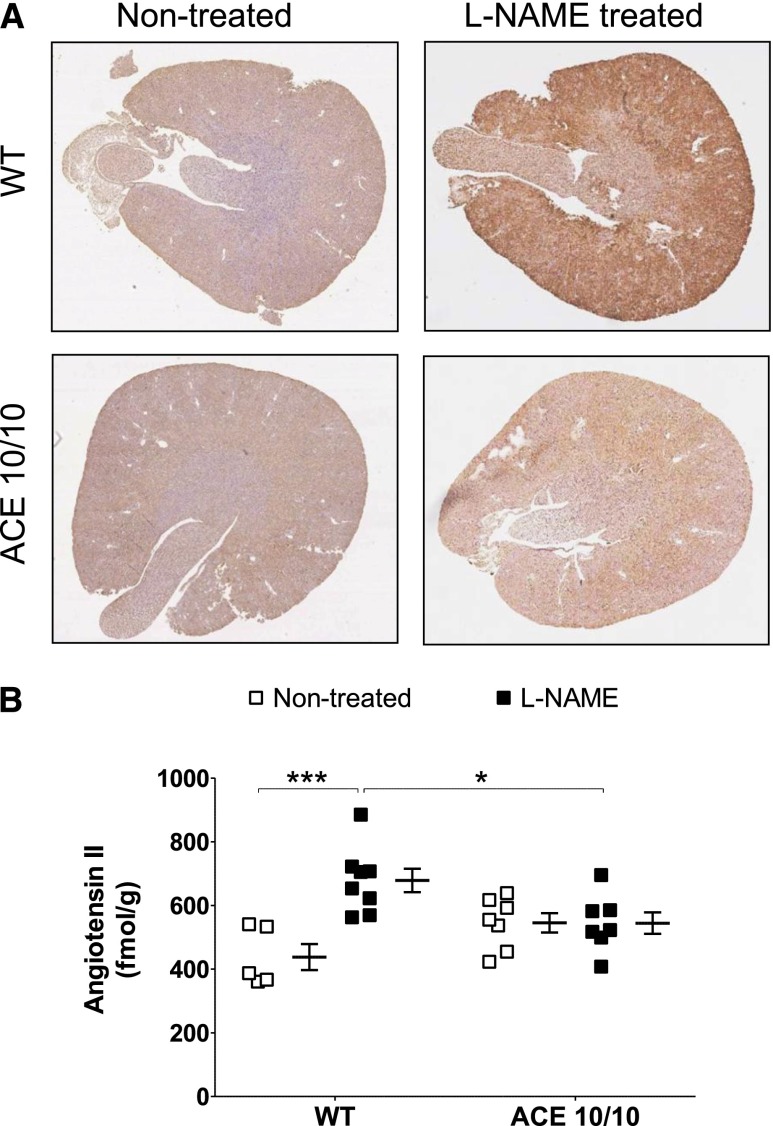
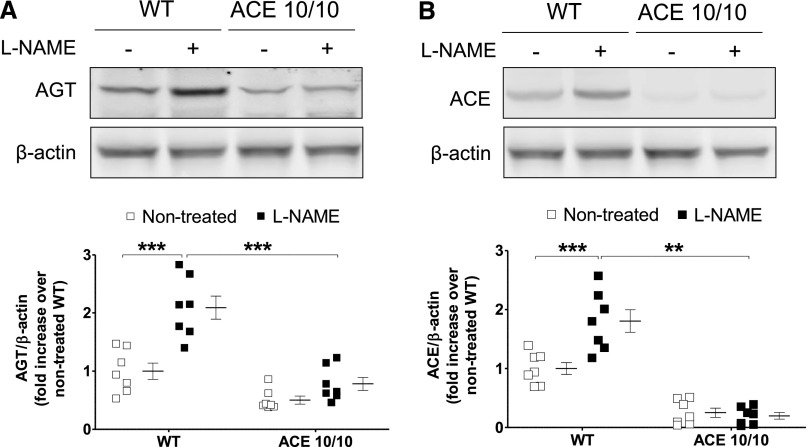
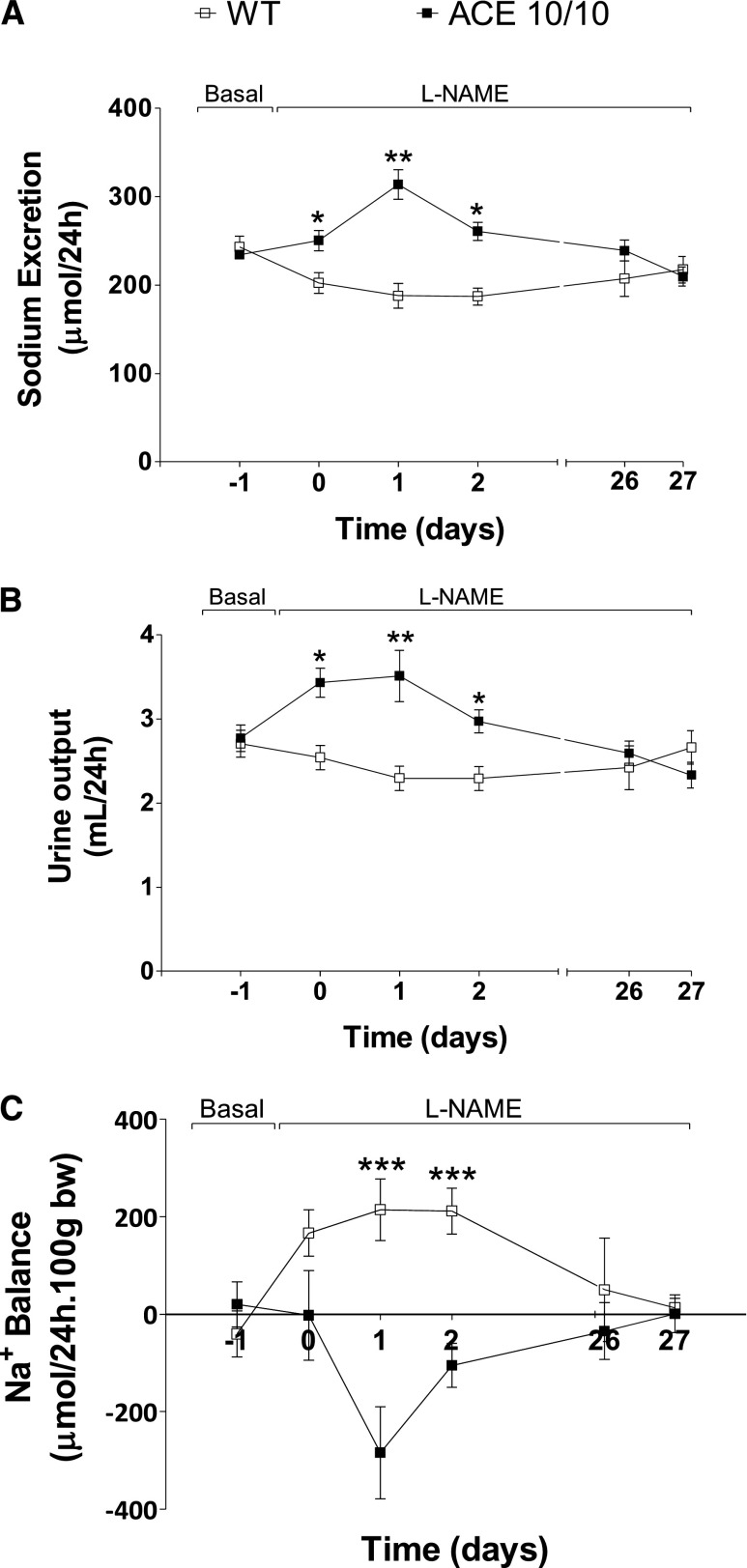
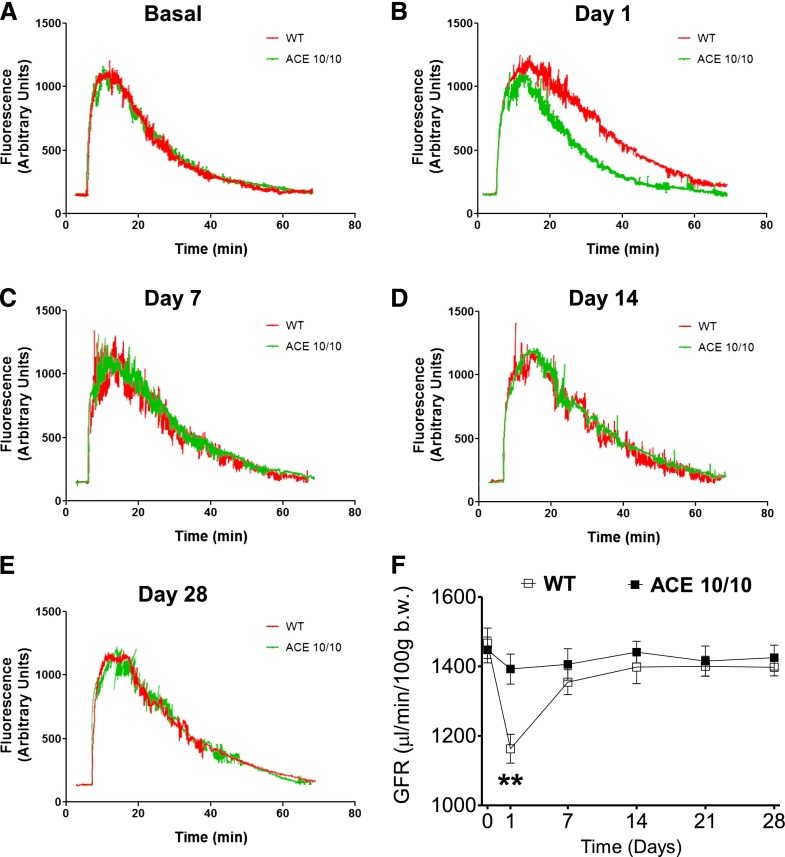
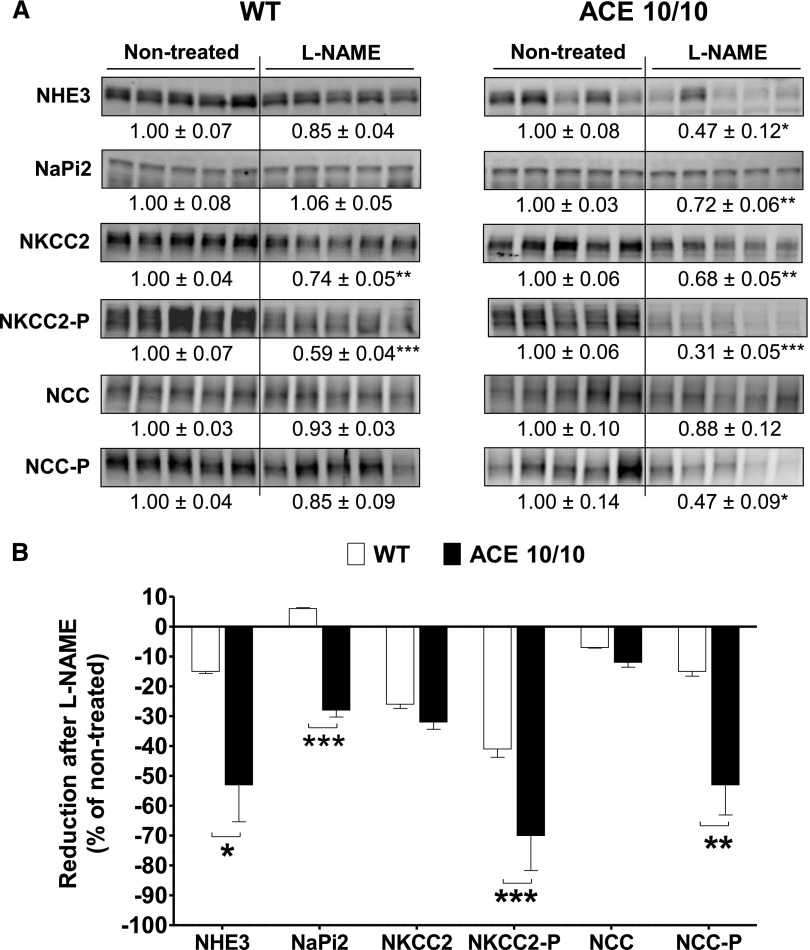
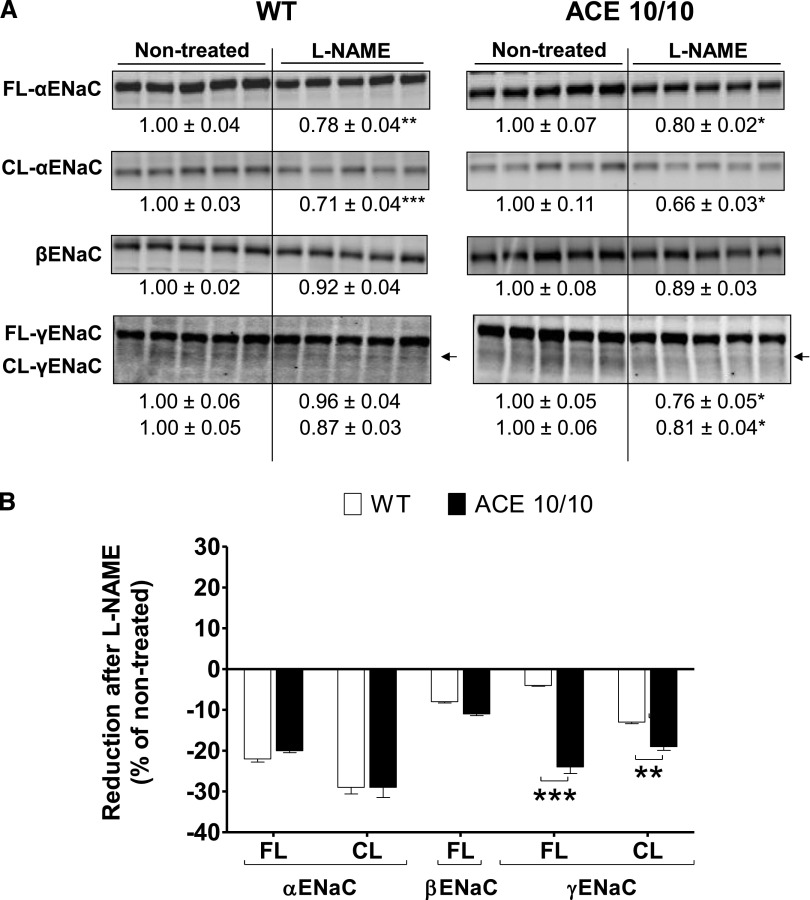
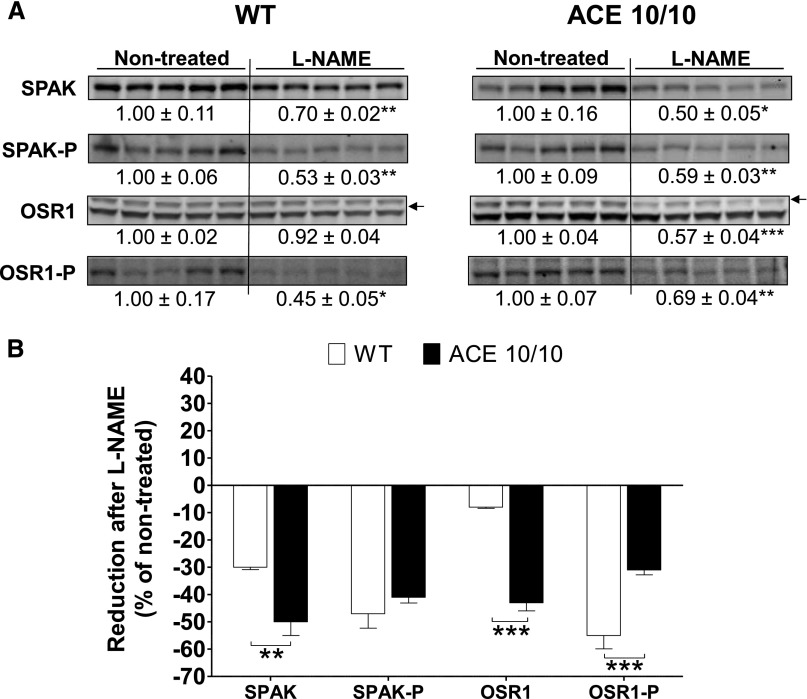
The kidney is an important source of angiotensin-converting enzyme (ACE) in many species, including humans. However, the specific effects of local ACE on renal function and, by extension, BP control are not completely understood. We previously showed that mice lacking renal ACE, are resistant to the hypertension induced by angiotensin II infusion. Here, we examined the responses of these mice to the low-systemic angiotensin II hypertensive model of nitric oxide synthesis inhibition with L-NAME. In contrast to wild-type mice, mice without renal ACE did not develop hypertension, had lower renal angiotensin II levels, and enhanced natriuresis in response to L-NAME. During L-NAME treatment, the absence of renal ACE was associated with blunted GFR responses; greater reductions in abundance of proximal tubule Na(+)/H(+) exchanger 3, Na(+)/Pi co-transporter 2, phosphorylated Na(+)/K(+)/Cl(-) cotransporter, and phosphorylated Na(+)/Cl(-) cotransporter; and greater reductions in abundance and processing of the γ isoform of the epithelial Na(+) channel. In summary, the presence of ACE in renal tissue facilitates angiotensin II accumulation, GFR reductions, and changes in the expression levels and post-translational modification of sodium transporters that are obligatory for sodium retention and hypertension in response to nitric oxide synthesis inhibition.
Keywords: ACE; L-NAME; hypertension; kidney; mice.
Copyright © 2014 by the American Society of Nephrology.
Figures
Comment in
-
Renal angiotensin-converting enzyme upregulation: a prerequisite for nitric oxide synthase inhibition-induced hypertension?J Am Soc Nephrol. 2014 Dec;25(12):2679-81. doi: 10.1681/ASN.2014060549. Epub 2014 Jul 10. J Am Soc Nephrol. 2014. PMID: 25012169 Free PMC article. No abstract available.
References
-
- Ng KK, Vane JR: Conversion of angiotensin I to angiotensin II. Nature 216: 762–766, 1967 - PubMed
-
- Alderman MH, Cohen HW, Sealey JE, Laragh JH: Plasma renin activity levels in hypertensive persons: Their wide range and lack of suppression in diabetic and in most elderly patients. Am J Hypertens 17: 1–7, 2004 - PubMed
-
- van den Meiracker AH, Man in ’t Veld AJ, Admiraal PJ, Ritsema van Eck HJ, Boomsma F, Derkx FH, Schalekamp MA: Partial escape of angiotensin converting enzyme (ACE) inhibition during prolonged ACE inhibitor treatment: Does it exist and does it affect the antihypertensive response? J Hypertens 10: 803–812, 1992 - PubMed
-
- Nussberger J, Brunner DB, Waeber B, Brunner HR: Plasma angiotensins under sustained converting enzyme inhibition with enalapril in normal humans. J Hypertens Suppl 3: S269–S270, 1985 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 DK083785/DK/NIDDK NIH HHS/United States
- T32 DK007770/DK/NIDDK NIH HHS/United States
- R56 DK083785/DK/NIDDK NIH HHS/United States
- P30 GM103337/GM/NIGMS NIH HHS/United States
- GM074771/GM/NIGMS NIH HHS/United States
- DK083785/DK/NIDDK NIH HHS/United States
- R01 GM074771/GM/NIGMS NIH HHS/United States
- R01 HL110353/HL/NHLBI NIH HHS/United States
- R03 DK101592/DK/NIDDK NIH HHS/United States
- 5T32DK007770-12/DK/NIDDK NIH HHS/United States
- HL110353/HL/NHLBI NIH HHS/United States
- R00-DK083455/DK/NIDDK NIH HHS/United States
- R00 DK083455/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
