

GIV/girdin links vascular endothelial growth factor signaling to Akt survival signaling in podocytes independent of nephrin
- PMID: 25012178
- PMCID: PMC4310647
- DOI: 10.1681/ASN.2013090985
GIV/girdin links vascular endothelial growth factor signaling to Akt survival signaling in podocytes independent of nephrin
Abstract
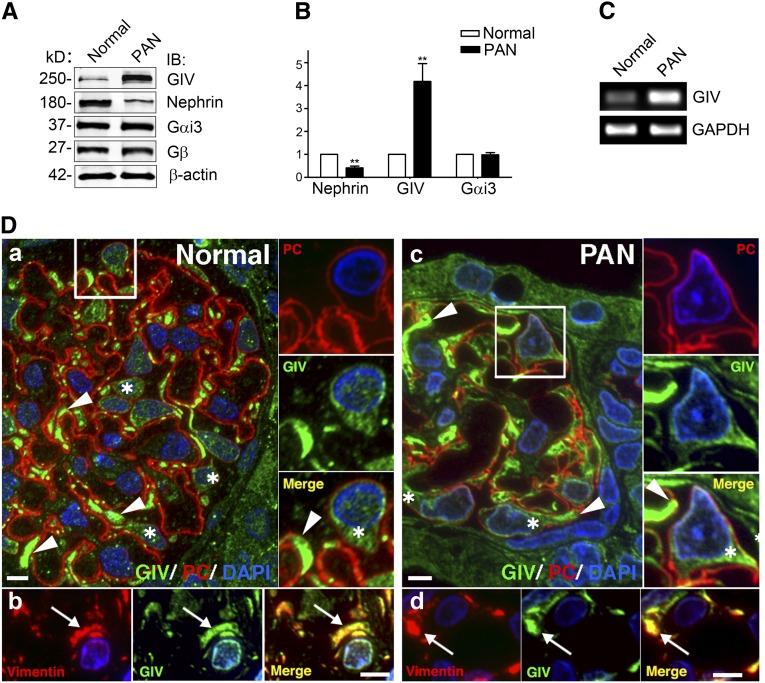
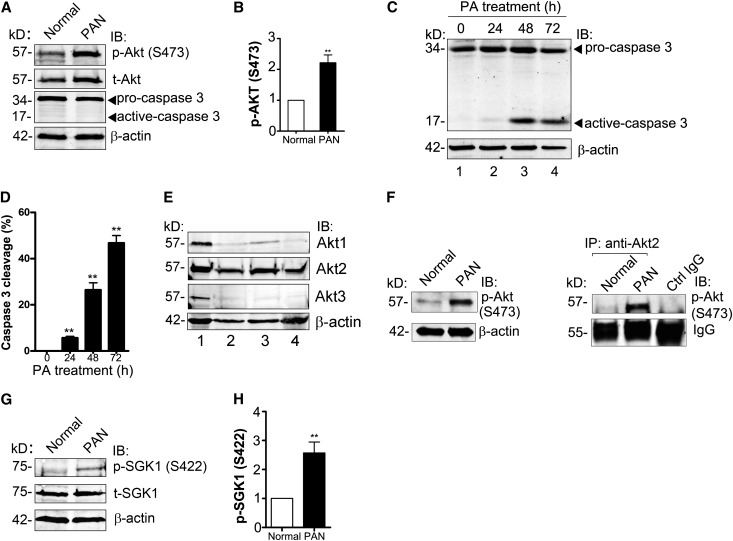
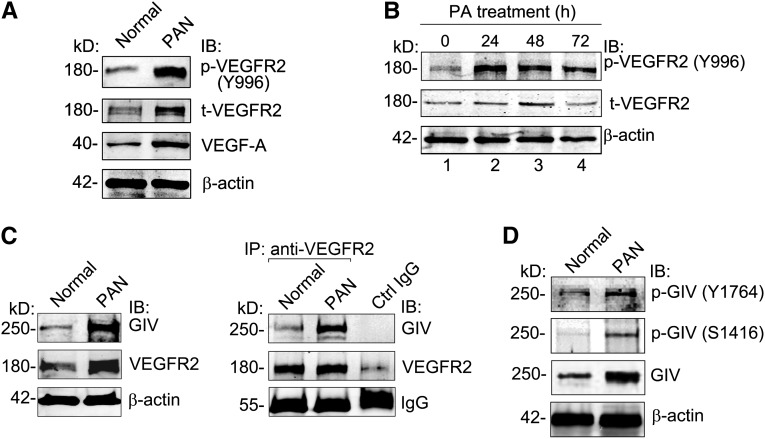
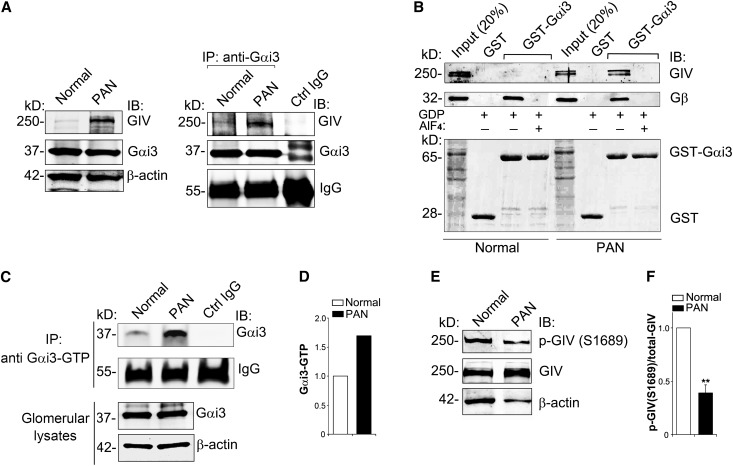
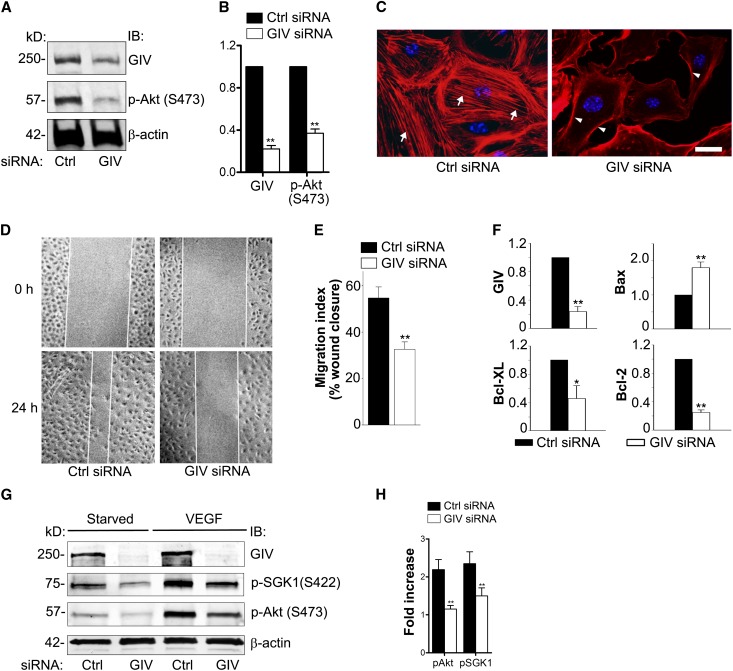
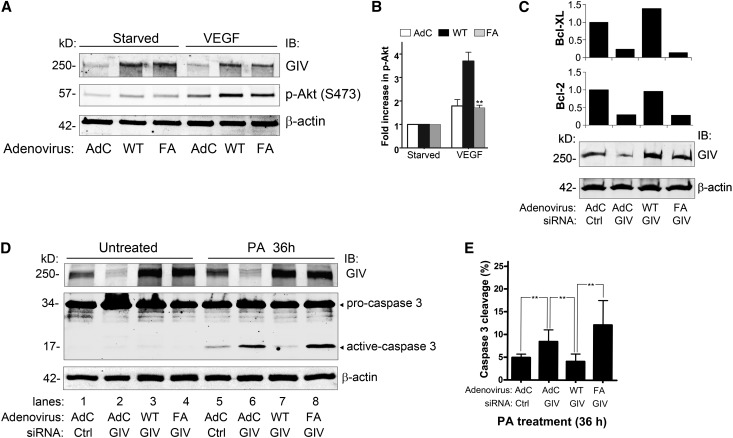
Podocytes are critically involved in the maintenance of the glomerular filtration barrier and are key targets of injury in many glomerular diseases. Chronic injury leads to progressive loss of podocytes, glomerulosclerosis, and renal failure. Thus, it is essential to maintain podocyte survival and avoid apoptosis after acute glomerular injury. In normal glomeruli, podocyte survival is mediated via nephrin-dependent Akt signaling. In several glomerular diseases, nephrin expression decreases and podocyte survival correlates with increased vascular endothelial growth factor (VEGF) signaling. How VEGF signaling contributes to podocyte survival and prevents apoptosis remains unknown. We show here that Gα-interacting, vesicle-associated protein (GIV)/girdin mediates VEGF receptor 2 (VEGFR2) signaling and compensates for nephrin loss. In puromycin aminonucleoside nephrosis (PAN), GIV expression increased, GIV was phosphorylated by VEGFR2, and p-GIV bound and activated Gαi3 and enhanced downstream Akt2, mammalian target of rapamycin complex 1 (mTORC1), and mammalian target of rapamycin complex-2 (mTORC2) signaling. In GIV-depleted podocytes, VEGF-induced Akt activation was abolished, apoptosis was triggered, and cell migration was impaired. These effects were reversed by introducing GIV but not a GIV mutant that cannot activate Gαi3. Our data indicate that after PAN injury, VEGF promotes podocyte survival by triggering assembly of an activated VEGFR2/GIV/Gαi3 signaling complex and enhancing downstream PI3K/Akt survival signaling. Because of its important role in promoting podocyte survival, GIV may represent a novel target for therapeutic intervention in the nephrotic syndrome and other proteinuric diseases.
Keywords: VEGF; cell survival; glomerular disease; podocyte.
Copyright © 2015 by the American Society of Nephrology.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
