

Class I and IIa histone deacetylases have opposite effects on sclerostin gene regulation
- PMID: 25012661
- PMCID: PMC4155667
- DOI: 10.1074/jbc.M114.564997
Class I and IIa histone deacetylases have opposite effects on sclerostin gene regulation
Abstract
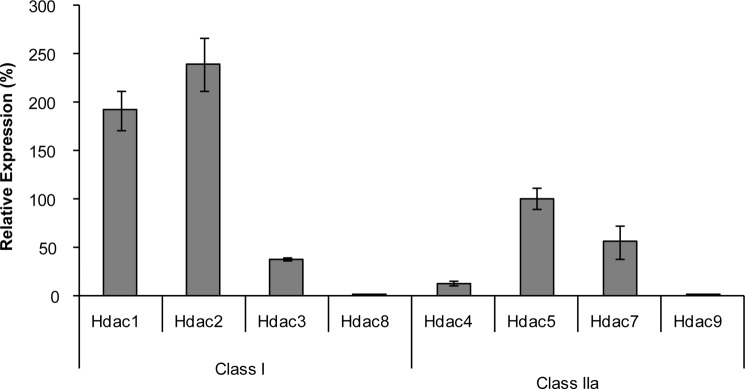
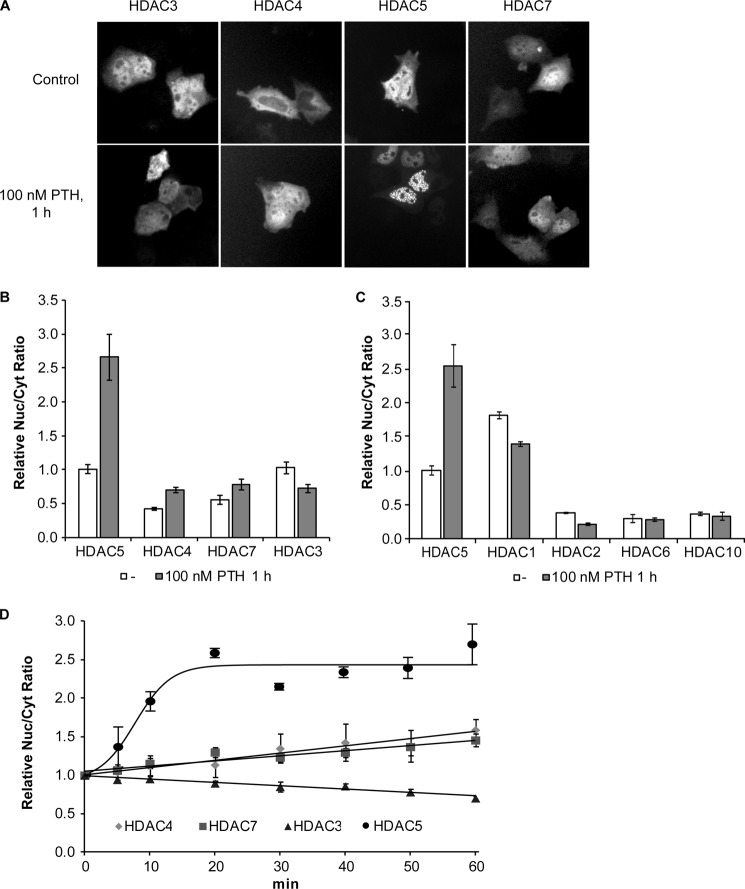
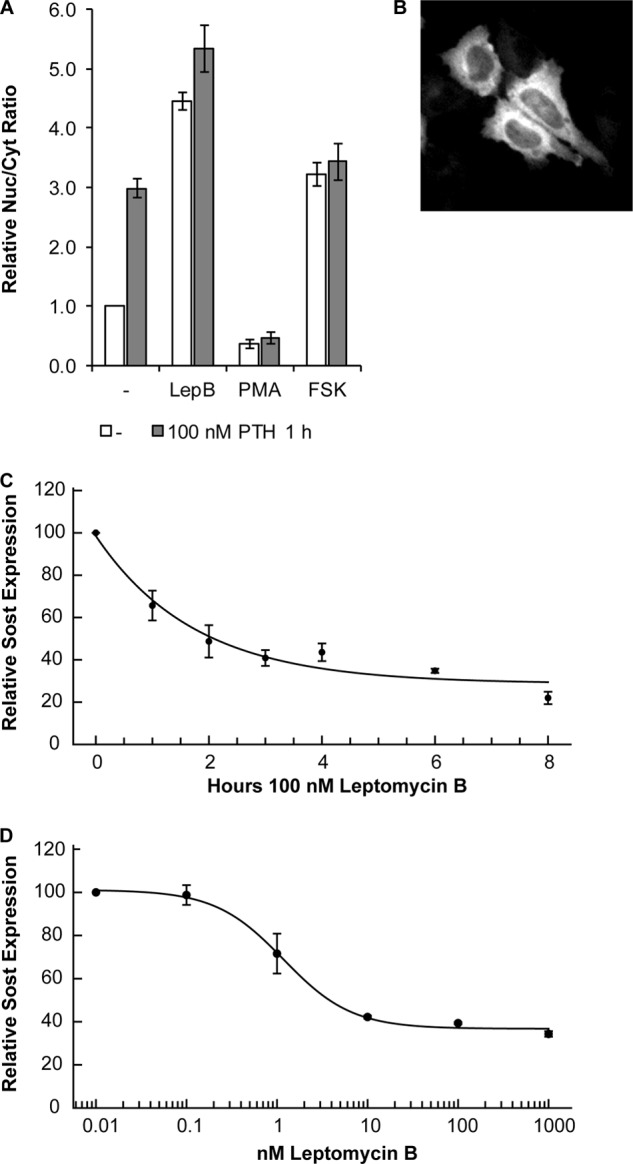
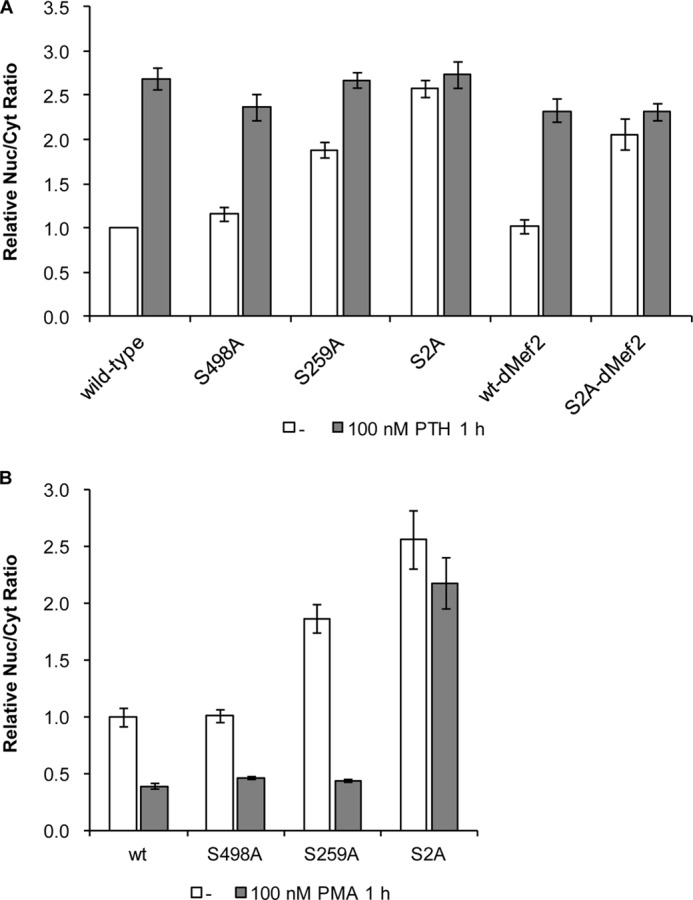
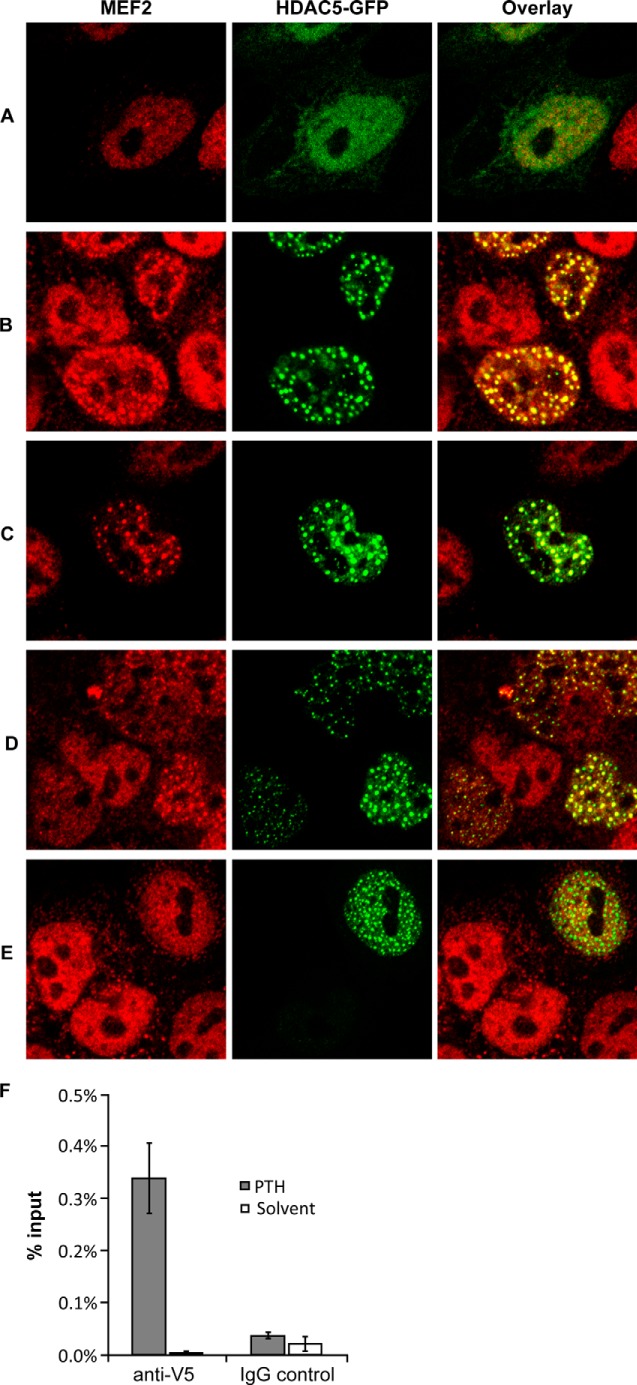
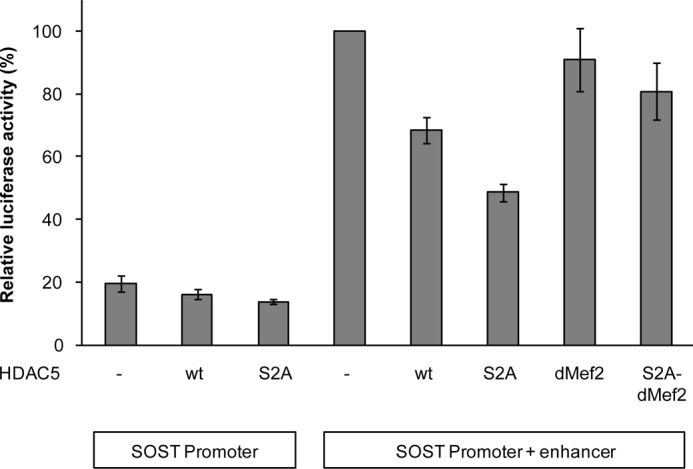
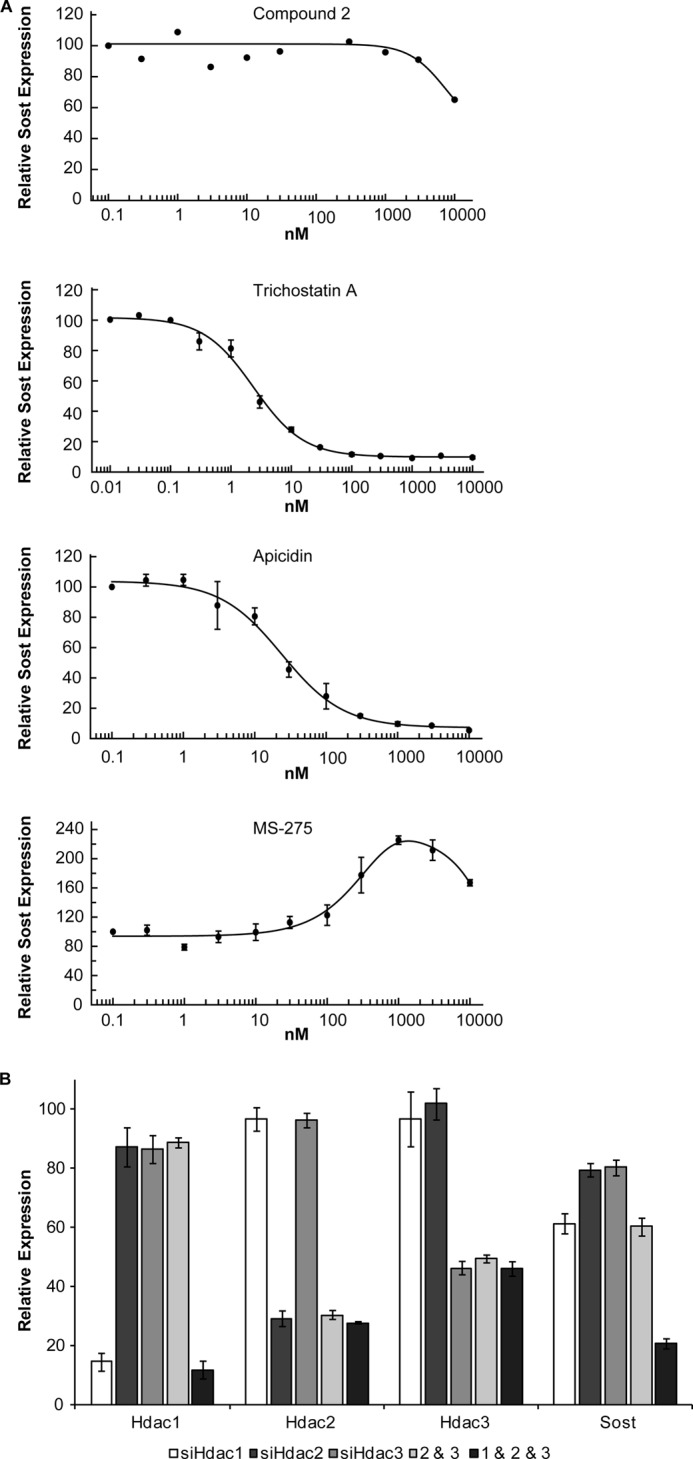
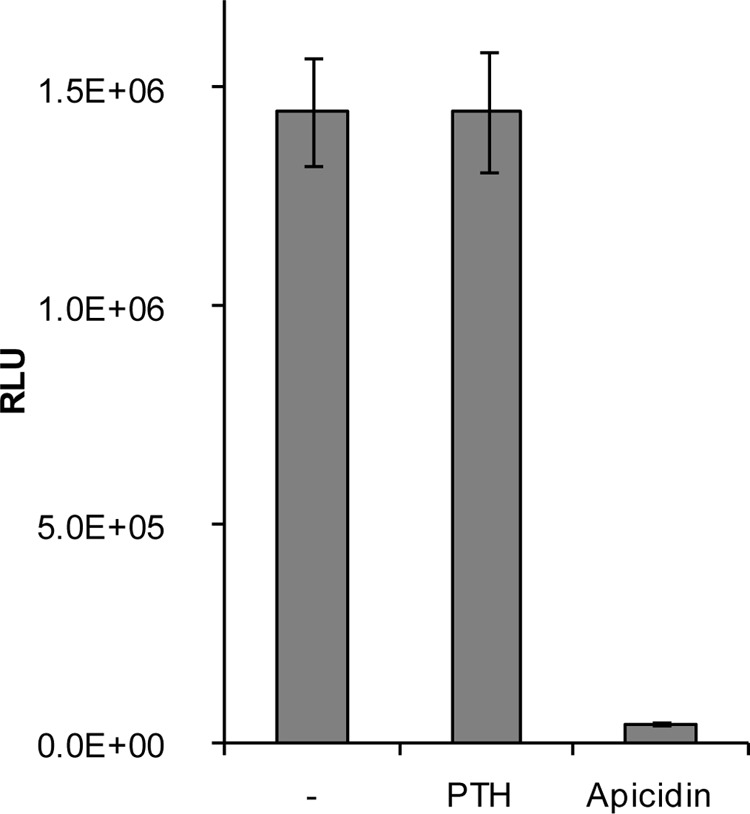
Adult bone mass is controlled by the bone formation repressor sclerostin (SOST). Previously, we have shown that intermittent parathyroid hormone (PTH) bone anabolic therapy involves SOST expression reduction by inhibiting myocyte enhancer factor 2 (MEF2), which activates a distant bone enhancer. Here, we extended our SOST gene regulation studies by analyzing a role of class I and IIa histone deacetylases (HDACs), which are known regulators of MEF2s. Expression analysis using quantitative PCR (qPCR) showed high expression of HDACs 1 and 2, lower amounts of HDACs 3, 5, and 7, low amounts of HDAC4, and no expression of HDACs 8 and 9 in constitutively SOST-expressing UMR106 osteocytic cells. PTH-induced Sost suppression was associated with specific rapid nuclear accumulation of HDAC5 and co-localization with MEF2s in nuclear speckles requiring serine residues 259 and 498, whose phosphorylations control nucleocytoplasmic shuttling. Increasing nuclear levels of HDAC5 in UMR106 by blocking nuclear export with leptomycin B (LepB) or overexpression in transient transfection assays inhibited endogenous Sost transcription and reporter gene expression, respectively. This repressor effect of HDAC5 did not require catalytic activity using specific HDAC inhibitors. In contrast, inhibition of class I HDAC activities and expression using RNA interference suppressed constitutive Sost expression in UMR106 cells. An unbiased comprehensive search for involved HDAC targets using an acetylome analysis revealed several non-histone proteins as candidates. These findings suggest that PTH-mediated Sost repression involves nuclear accumulation of HDAC inhibiting the MEF2-dependent Sost bone enhancer, and class I HDACs are required for constitutive Sost expression in osteocytes.
Keywords: Gene Regulation; Histone Deacetylase (HDAC); Parathyroid Hormone; Sclerostin; Transcription Corepressor; Transcriptional Coactivator.
© 2014 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Li X., Ominsky M. S., Niu Q. T., Sun N., Daugherty B., D'Agostin D., Kurahara C., Gao Y., Cao J., Gong J., Asuncion F., Barrero M., Warmington K., Dwyer D., Stolina M., Morony S., Sarosi I., Kostenuik P. J., Lacey D. L., Simonet W. S., Ke H. Z., Paszty C. (2008) Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J. Bone Miner. Res. 23, 860–869 - PubMed
-
- Winkler D. G., Sutherland M. K., Geoghegan J. C., Yu C., Hayes T., Skonier J. E., Shpektor D., Jonas M., Kovacevich B. R., Staehling-Hampton K., Appleby M., Brunkow M. E., Latham J. A. (2003) Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 22, 6267–6276 - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
