

Evolutionary and sequence-based relationships in bacterial AdoMet-dependent non-coding RNA methyltransferases
- PMID: 25012753
- PMCID: PMC4119055
- DOI: 10.1186/1756-0500-7-440
Evolutionary and sequence-based relationships in bacterial AdoMet-dependent non-coding RNA methyltransferases
Abstract
Background: RNA post-transcriptional modification is an exciting field of research that has evidenced this editing process as a sophisticated epigenetic mechanism to fine tune the ribosome function and to control gene expression. Although tRNA modifications seem to be more relevant for the ribosome function and cell physiology as a whole, some rRNA modifications have also been seen to play pivotal roles, essentially those located in central ribosome regions. RNA methylation at nucleobases and ribose moieties of nucleotides appear to frequently modulate its chemistry and structure. RNA methyltransferases comprise a superfamily of highly specialized enzymes that accomplish a wide variety of modifications. These enzymes exhibit a poor degree of sequence similarity in spite of using a common reaction cofactor and modifying the same substrate type.
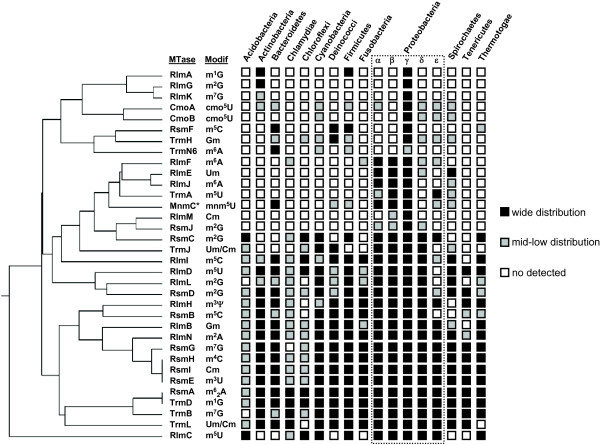
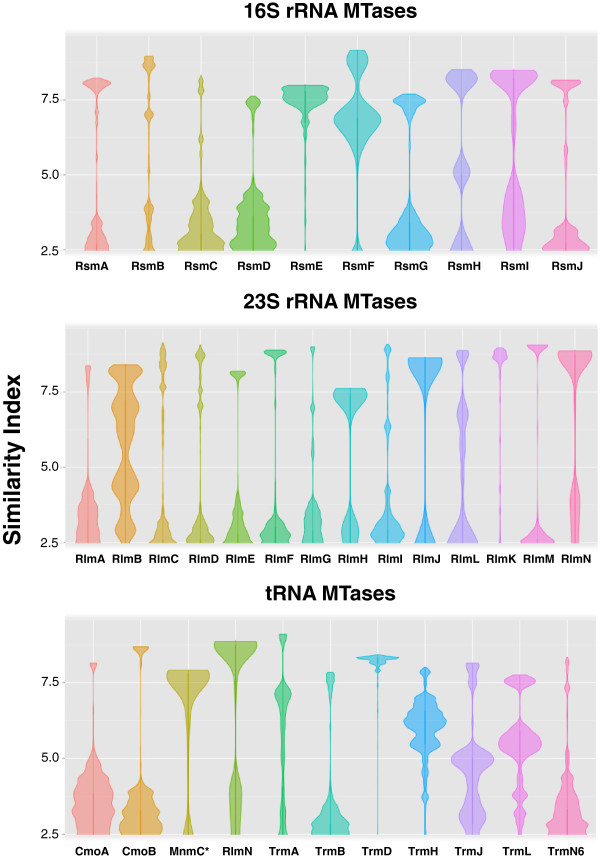
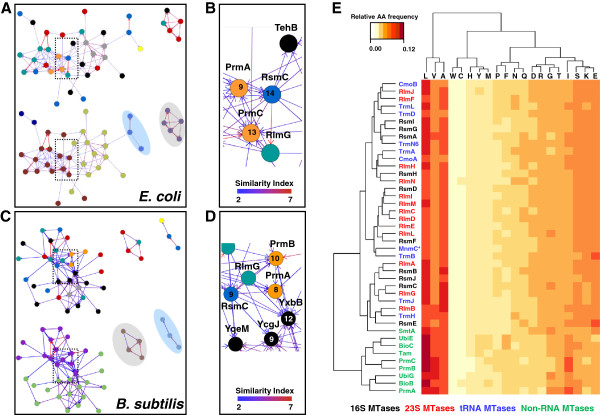
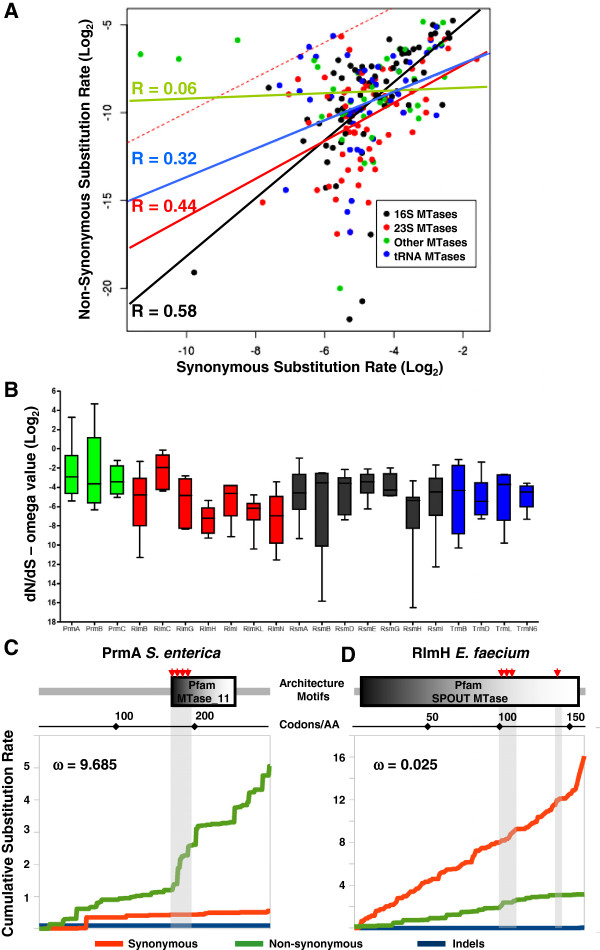
Results: Relationships and lineages of RNA methyltransferases have been extensively discussed, but no consensus has been reached. To shed light on this topic, we performed amino acid and codon-based sequence analyses to determine phylogenetic relationships and molecular evolution. We found that most Class I RNA MTases are evolutionarily related to protein and cofactor/vitamin biosynthesis methyltransferases. Additionally, we found that at least nine lineages explain the diversity of RNA MTases. We evidenced that RNA methyltransferases have high content of polar and positively charged amino acid, which coincides with the electrochemistry of their substrates.
Conclusions: After studying almost 12,000 bacterial genomes and 2,000 patho-pangenomes, we revealed that molecular evolution of Class I methyltransferases matches the different rates of synonymous and non-synonymous substitutions along the coding region. Consequently, evolution on Class I methyltransferases selects against amino acid changes affecting the structure conformation.
Figures
References
-
- Grosjean H. In: Topics in Current Genetics. 12. Hohmann S, editor. New York: Springer Verlag; 2005. Fine tuning of RNA functions by modification and editing.
-
- Decatur WA, Fournier MJ. rRNA modifications and ribosome function. Trends Biochem Sci. 2002;27(7):344–351. - PubMed
-
- Björk GR, Hagervall TG. In: EcoSal—Escherichia coli and Salmonella: cellular and molecular biology. Böck RCI, Kaper JB, Neidhardt FC, Nyström T, Rudd KE, Squires CL, editor. Washington, D.C: ASM Press; 2005. Transfer RNA modification.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
