

A large-scale RNAi-based mouse tumorigenesis screen identifies new lung cancer tumor suppressors that repress FGFR signaling
- PMID: 25015643
- PMCID: PMC4184919
- DOI: 10.1158/2159-8290.CD-13-0747
A large-scale RNAi-based mouse tumorigenesis screen identifies new lung cancer tumor suppressors that repress FGFR signaling
Abstract
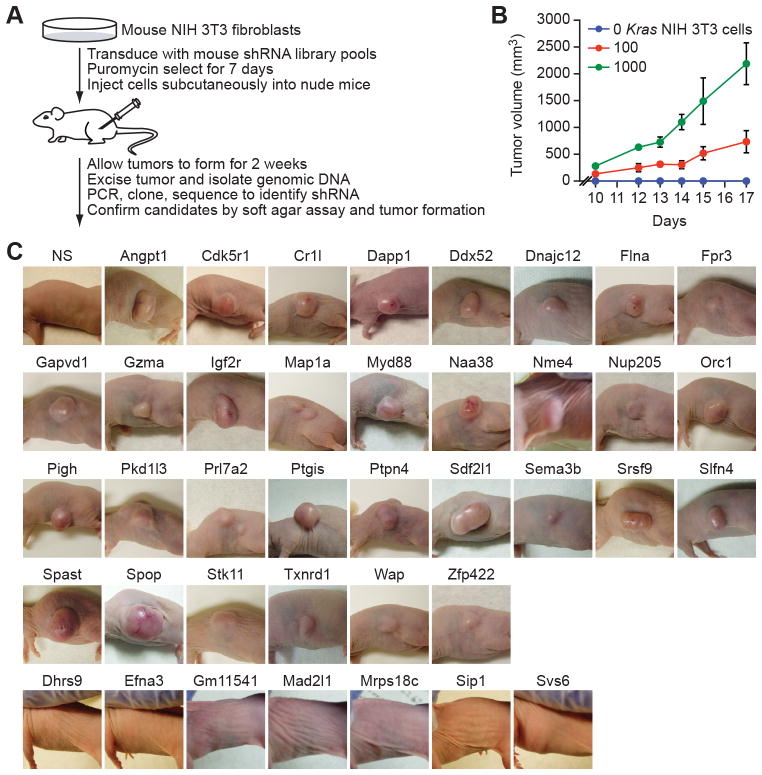
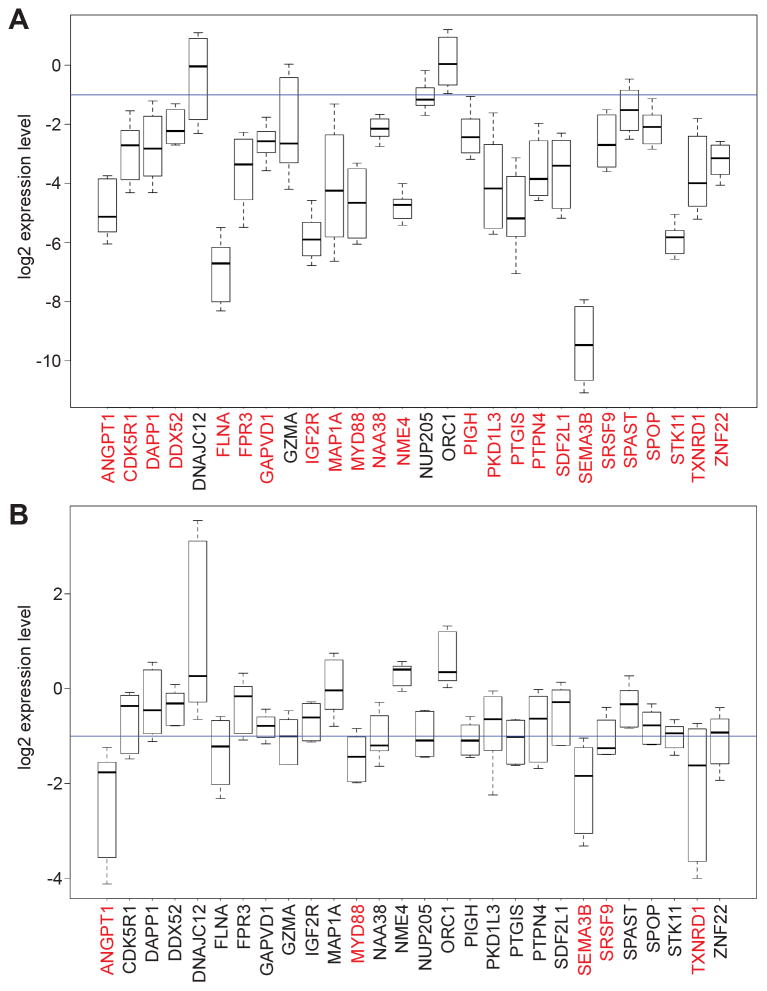
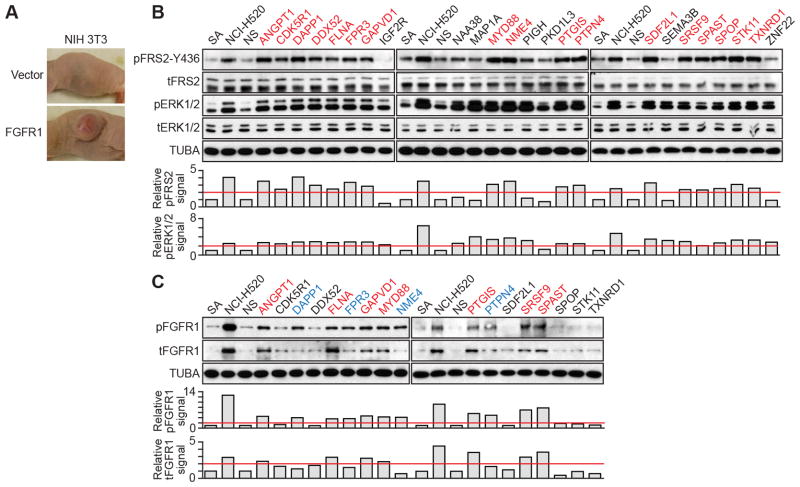
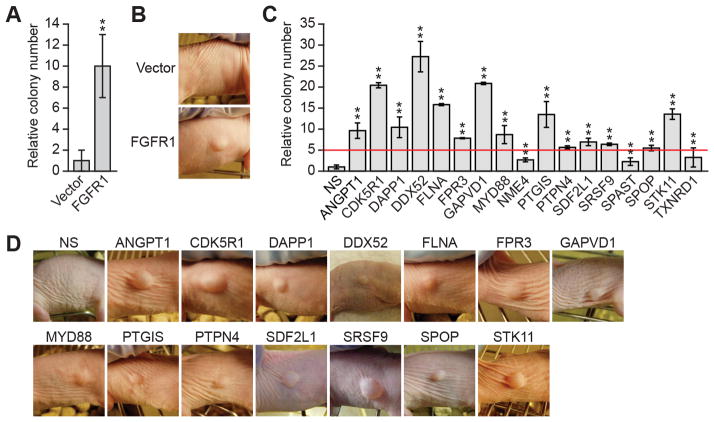
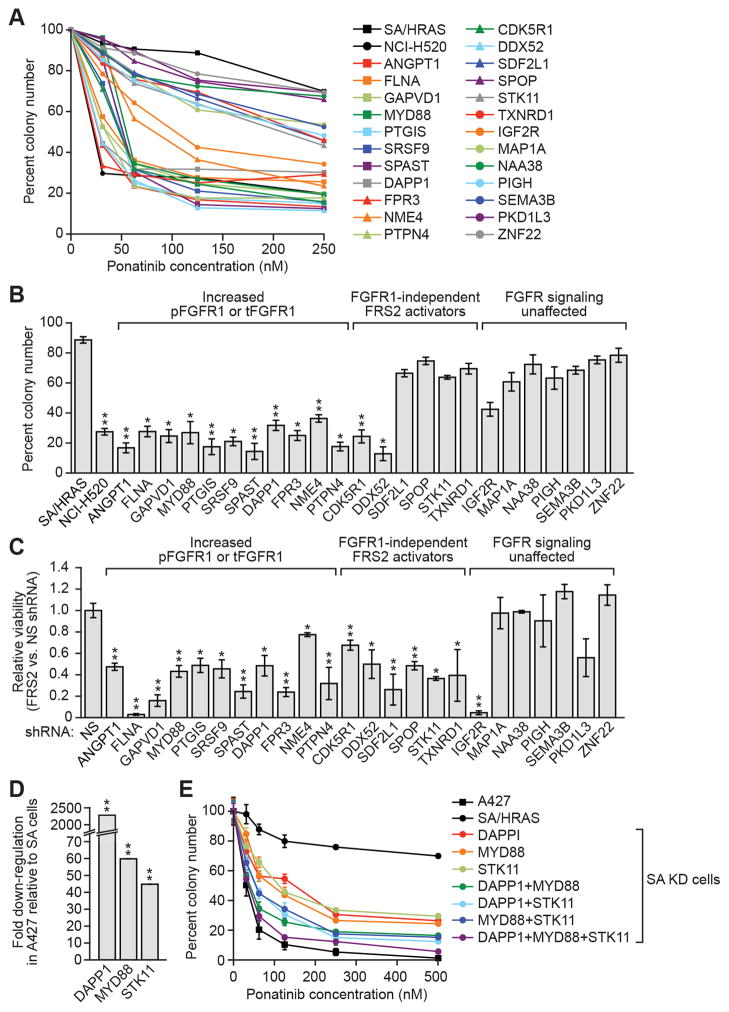
To discover new tumor-suppressor genes (TSG), we developed a functional genomics approach in which immortalized but nontumorigenic cells were stably transduced with large-scale shRNA pools and tested for tumor formation in mice. Identification of shRNAs in resulting tumors revealed candidate TSGs, which were validated experimentally and by analyzing expression in human tumor samples. Using this approach, we identified 24 TSGs that were significantly downregulated in human lung squamous cell carcinomas (hLSCC). Amplification of fibroblast growth factor receptor 1 (FGFR1), which aberrantly increases FGFR signaling, is a common genetic alteration in hLSCCs. Remarkably, we found that 17 of the TSGs encode repressors of FGFR signaling. Knockdown of 14 of these TSGs transformed immortalized human bronchial epithelial cells and, in most cases, rendered them sensitive to FGFR inhibitors. Our results indicate that increased FGFR signaling promotes tumorigenesis in many hLSCCs that lack FGFR1 amplification or activating mutations.
Significance: A functional genomics approach identifies new lung TSGs whose loss aberrantly increases FGFR signaling to promote tumorigenesis. These TSGs are frequently downregulated in hLSCCs, indicating that increased FGFR signaling promotes tumorigenesis in many hLSCCs lacking FGFR1 amplification or activating mutations.
©2014 American Association for Cancer Research.
Conflict of interest statement
The authors disclose no potential conflicts of interest.
No potential conflicts of interest were disclosed by the authors.
Figures

References
-
- Loeb LA. A mutator phenotype in cancer. Cancer Res. 2001;61:3230–9. - PubMed
-
- Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet. 2007;16(Spec No 1):R50–9. - PubMed
-
- Stacey DW, Kung HF. Transformation of NIH 3T3 cells by microinjection of Ha-ras p21 protein. Nature. 1984;310:508–11. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
