

Individual exome analysis in diagnosis and management of paediatric liver failure of indeterminate aetiology
- PMID: 25016221
- PMCID: PMC4203706
- DOI: 10.1016/j.jhep.2014.06.038
Individual exome analysis in diagnosis and management of paediatric liver failure of indeterminate aetiology
Abstract
Background & aims: In children with liver failure, as many as half remain of indeterminate aetiology. This hinders timely consideration of optimal treatment options. We posit that a significant subset of these children harbour known inherited metabolic liver diseases with atypical presentation or novel inborn errors of metabolism. We investigated the utility of whole-exome sequencing in three children with advanced liver disease of indeterminate aetiology.
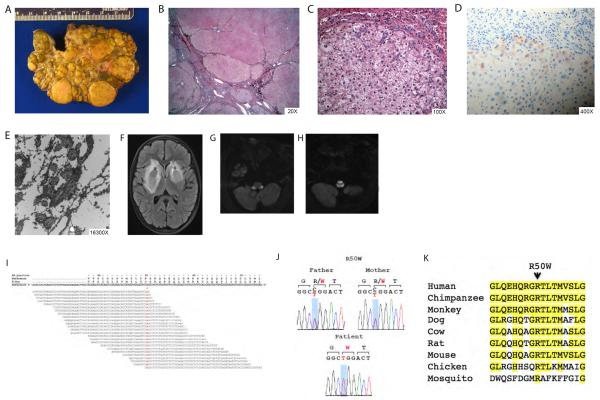
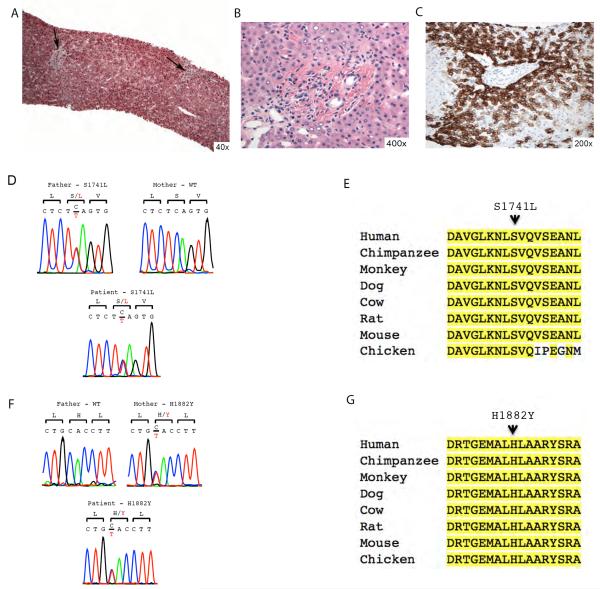
Methods: Patient 1 was a 10 year-old female diagnosed with Wilson disease but no detectable ATP7B mutations, and decompensated liver cirrhosis who underwent liver transplant and subsequently developed onset of neurodegenerative disease. Patient 2 was a full-term 2 day-old male with fatal acute liver failure of indeterminate aetiology. Patient 3 was an 8 year-old female with progressive syndromic cholestasis of unknown aetiology since age 3 months.
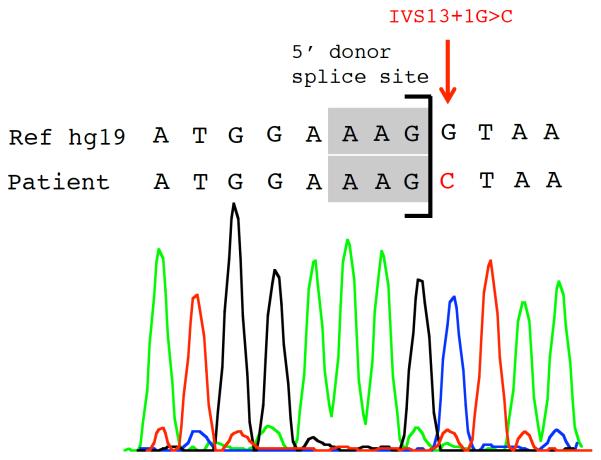
Results: Unbiased whole-exome sequencing of germline DNA revealed homozygous mutations in MPV17 and SERAC1 as the disease causing genes in patient 1 and 2, respectively. This is the first demonstration of SERAC1 loss-of-function associated fatal acute liver failure. Patient 1 expands the phenotypic spectrum of the MPV17-related hepatocerebral mitochondrial DNA depletion syndrome. Patient 3 was found to have syndromic cholestasis due to bi-allelic NOTCH2 mutations.
Conclusions: Our findings validate the application of whole-exome sequencing in the diagnosis and management of children with advanced liver disease of indeterminate aetiology, with the potential to enhance optimal selection of treatment options and adequate counselling of families. Moreover, whole-exome sequencing revealed a hitherto unrecognized phenotypic spectrum of inherited metabolic liver diseases.
Keywords: Genetic diagnosis; Germline mutations; Inherited metabolic liver diseases; Liver failure of indeterminate aetiology; Whole-exome sequencing.
Copyright © 2014 European Association for the Study of the Liver. Published by Elsevier B.V. All rights reserved.
Figures
References
-
- Jacob HJ, Abrams K, Bick DP, Brodie K, Dimmock DP, Farrell M, et al. Genomics in clinical practice: lessons from the front lines. Science translational medicine. 2013;5 194cm195. - PubMed
-
- Wortmann SB, Vaz FM, Gardeitchik T, Vissers LE, Renkema GH, Schuurs-Hoeijmakers JH, et al. Mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness. Nature genetics. 2012;44:797–802. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
