

Sudden unexpected fatal encephalopathy in adults with OTC gene mutations-Clues for early diagnosis and timely treatment
- PMID: 25026867
- PMCID: PMC4304088
- DOI: 10.1186/s13023-014-0105-9
Sudden unexpected fatal encephalopathy in adults with OTC gene mutations-Clues for early diagnosis and timely treatment
Abstract
Background: X-linked Ornithine Transcarbamylase deficiency (OTCD) is often unrecognized in adults, as clinical manifestations are non-specific, often episodic and unmasked by precipitants, and laboratory findings can be normal outside the acute phase. It may thus be associated with significant mortality if not promptly recognized and treated. The aim of this study was to provide clues for recognition of OTCD in adults and analyze the environmental factors that, interacting with OTC gene mutations, might have triggered acute clinical manifestations.
Methods: We carried out a clinical, biochemical and molecular study on five unrelated adult patients (one female and four males) with late onset OTCD, who presented to the Emergency Department (ED) with initial fatal encephalopathy. The molecular study consisted of OTC gene sequencing in the probands and family members and in silico characterization of the newly detected mutations.
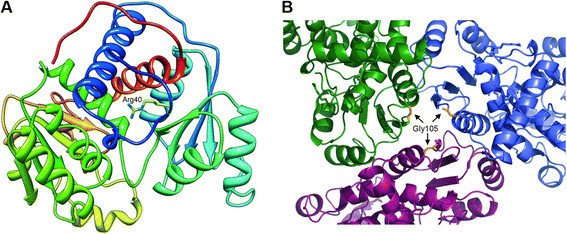
Results: We identified two new, c.119G>T (p.Arg40Leu) and c.314G>A (p.Gly105Glu), and three known OTC mutations. Both new mutations were predicted to cause a structural destabilization, correlating with late onset OTCD. We also identified, among the family members, 8 heterozygous females and 2 hemizygous asymptomatic males. Patients' histories revealed potential environmental triggering factors, including steroid treatment, chemotherapy, diet changes and hormone therapy for in vitro fertilization.
Conclusions: This report raises awareness of the ED medical staff in considering OTCD in the differential diagnosis of sudden neurological and behavioural disorders associated with hyperammonemia at any age and in both genders. It also widens the knowledge about combined effect of genetic and environmental factors in determining the phenotypic expression of OTCD.
Figures
References
-
- Brusilow SWHA. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver CR, Beaudet AL, Valle D, Sly WS, editor. McGraw-Hill, New York; 2001. Urea Cycle Enzymes; pp. 1909–1963.
-
- Numata S, Koda Y, Ihara K, Sawada T, Okano Y, Matsuura T, Endo F, Yoo HW, Arranz JA, Rubio V, Wermuth B, Ah Mew N, Tuchman M, Pinner JR, Kirk EP, Yoshino M. Mutant alleles associated with late-onset ornithine transcarbamylase deficiency in male patients have recurrently arisen and have been retained in some populations. J Hum Genet. 2010;55:18–22. doi: 10.1038/jhg.2009.113. - DOI - PubMed
-
- Ruegger CM, Lindner M, Ballhausen D, Baumgartner MR, Beblo S, Das A, Gautschi M, Glahn EM, Grunert SC, Hennermann J, Hochuli M, Huemer M, Karall D, Kölker S, Lachmann RH, Lotz-Havla A, Möslinger D, Nuoffer JM, Plecko B, Rutsch F, Santer R, Spiekerkoetter U, Staufner C, Stricker T, Wijburg FA, Williams M, Burgard P, Häberle J. Cross-sectional observational study of 208 patients with non-classical urea cycle disorders. J Inherit Metab Dis. 2014;37:21–30. doi: 10.1007/s10545-013-9624-0. - DOI - PMC - PubMed
-
- Haberle J, Boddaert N, Burlina A, Chakrapani A, Dixon M, Huemer M, Karall D, Martinelli D, Crespo PS, Santer R, Servais A, Valayannopoulos V, Lindner M, Rubio V, Dionisi-Vici C. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:32. doi: 10.1186/1750-1172-7-32. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
