

RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP)
- PMID: 25028896
- PMCID: PMC4259394
- DOI: 10.1038/nmeth.3029
RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP)
Abstract
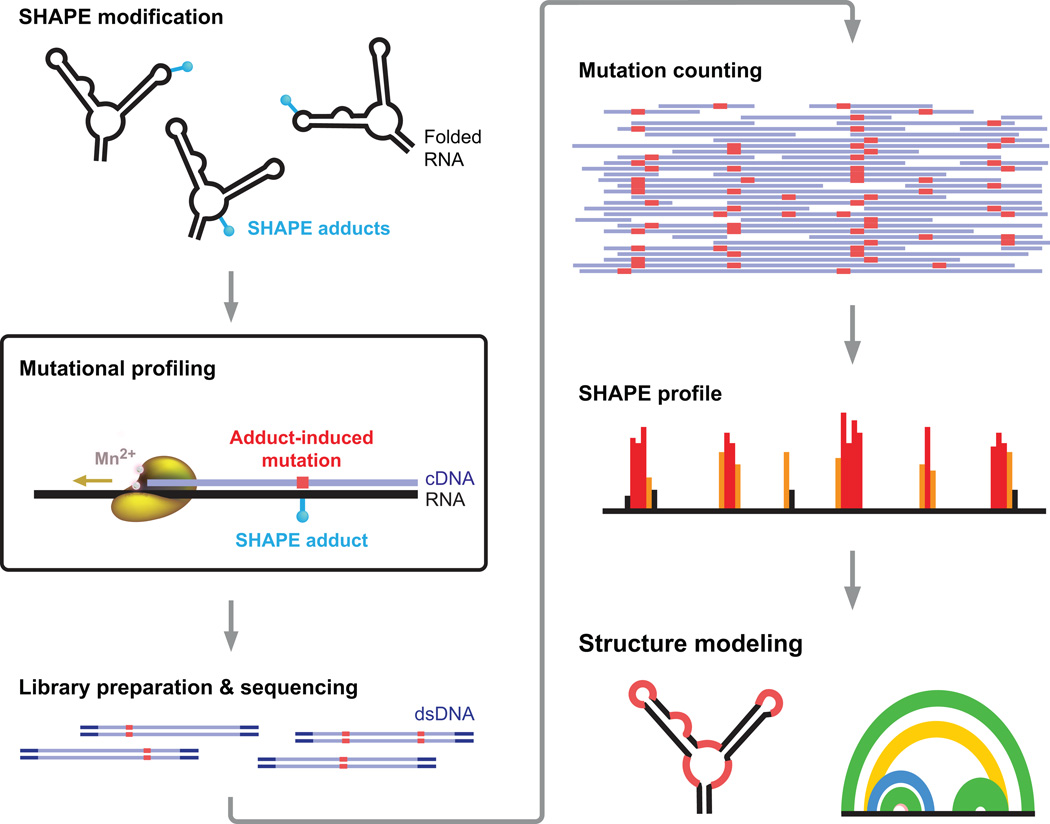
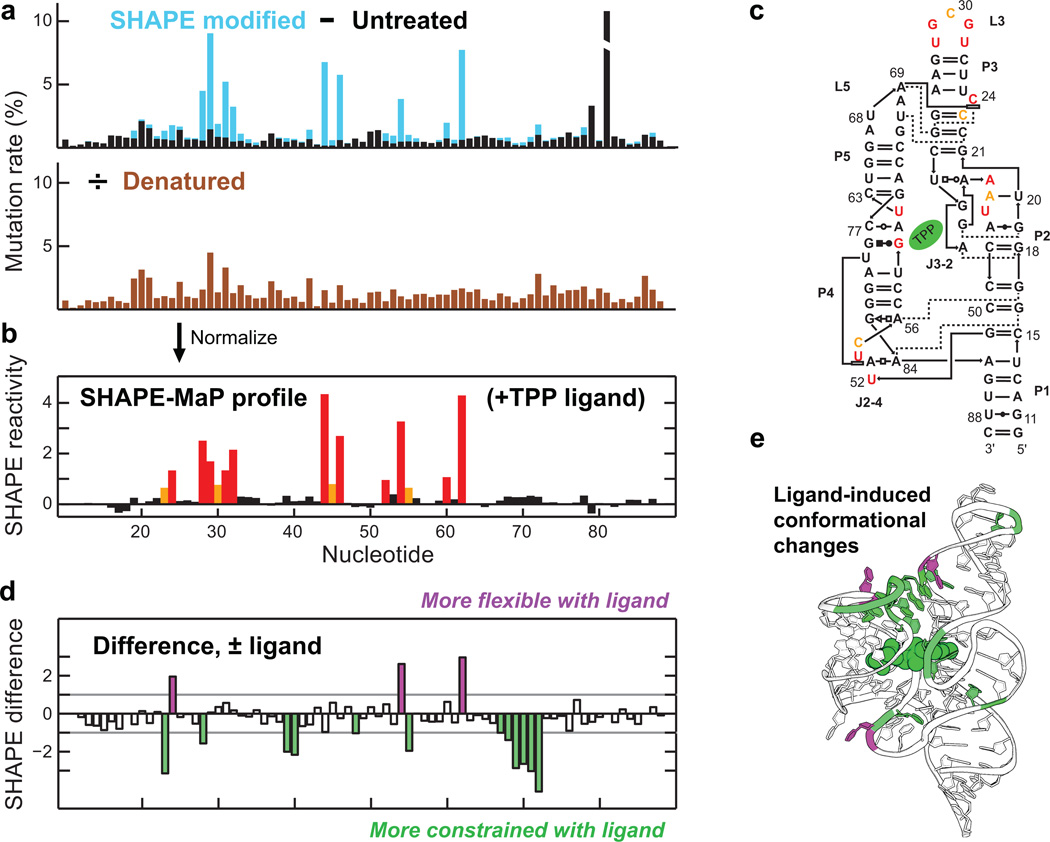
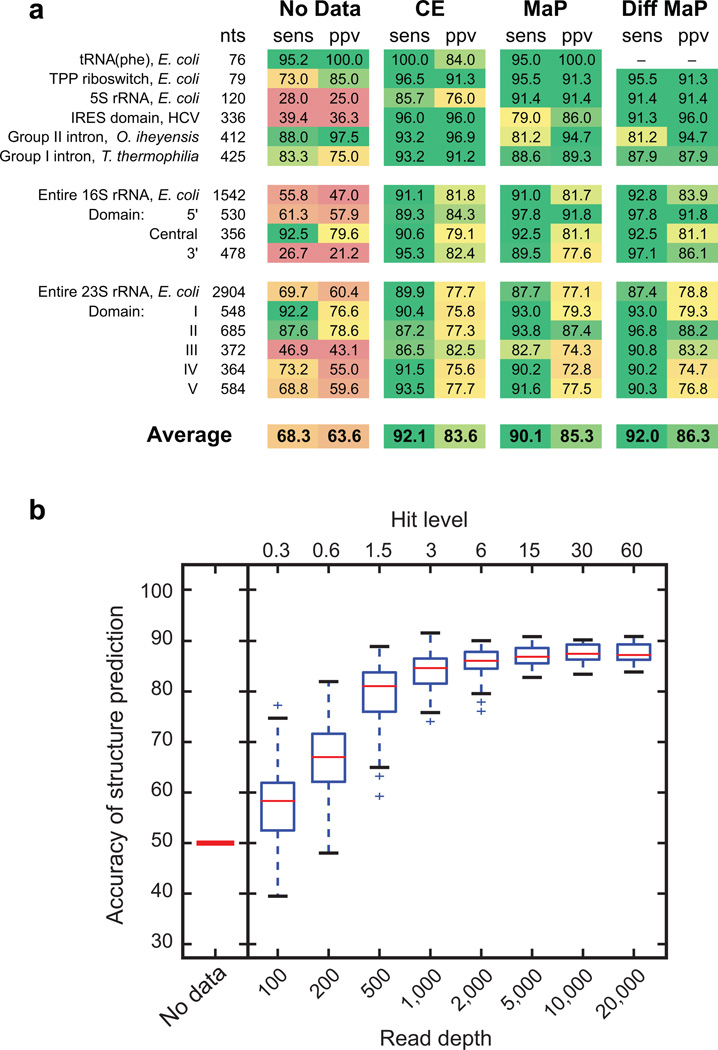
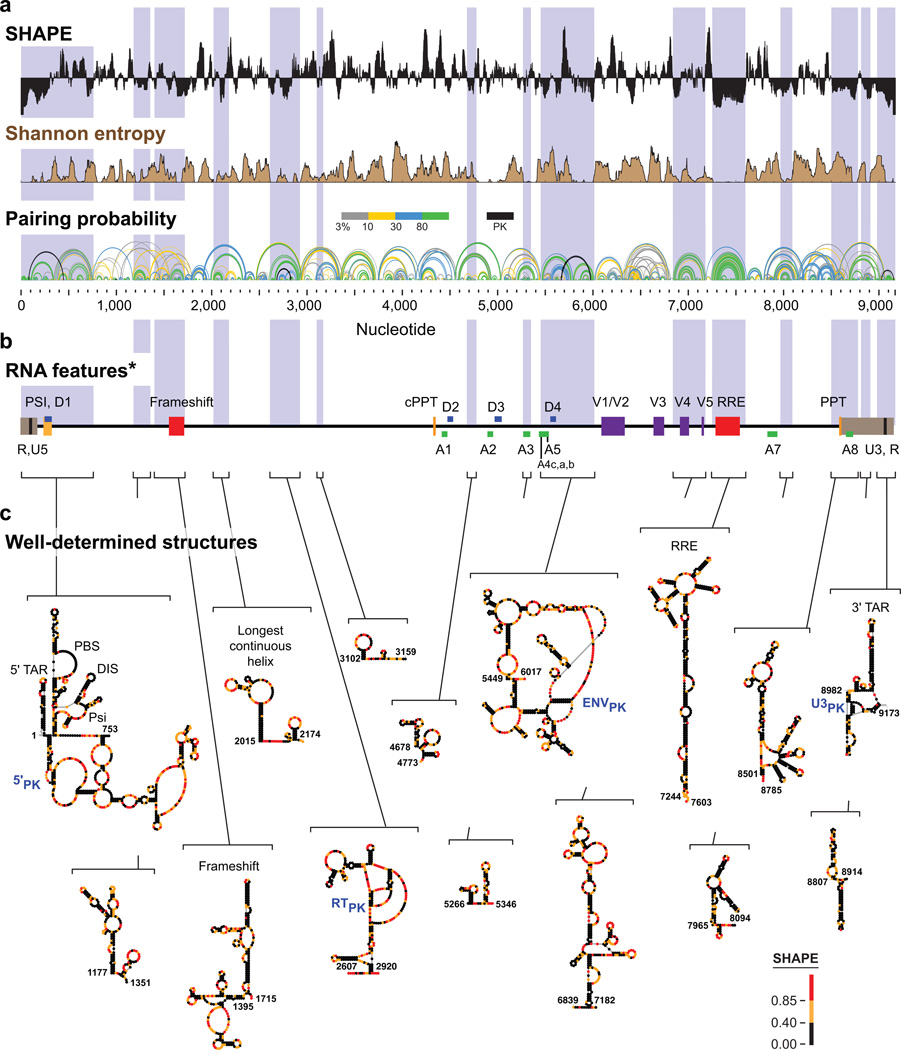
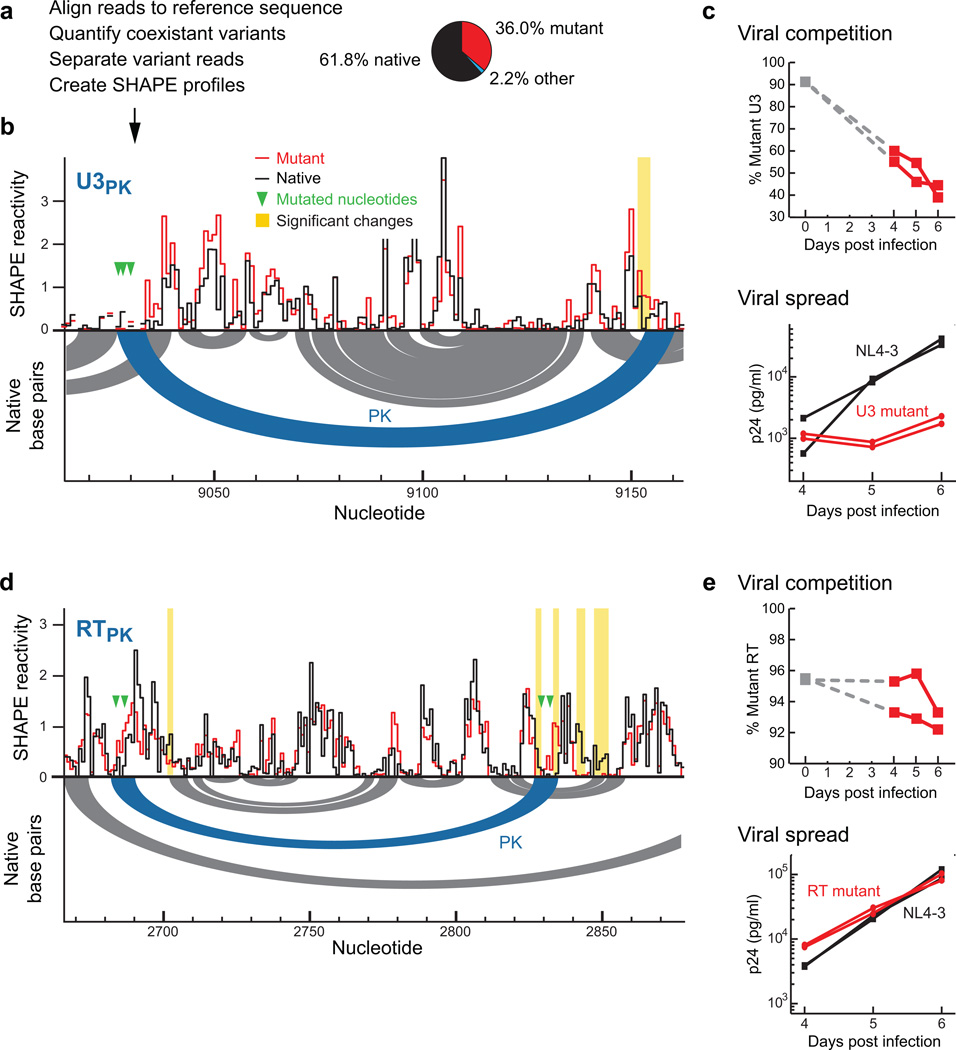
Many biological processes are RNA-mediated, but higher-order structures for most RNAs are unknown, which makes it difficult to understand how RNA structure governs function. Here we describe selective 2'-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) that makes possible de novo and large-scale identification of RNA functional motifs. Sites of 2'-hydroxyl acylation by SHAPE are encoded as noncomplementary nucleotides during cDNA synthesis, as measured by massively parallel sequencing. SHAPE-MaP-guided modeling identified greater than 90% of accepted base pairs in complex RNAs of known structure, and we used it to define a new model for the HIV-1 RNA genome. The HIV-1 model contains all known structured motifs and previously unknown elements, including experimentally validated pseudoknots. SHAPE-MaP yields accurate and high-resolution secondary-structure models, enables analysis of low-abundance RNAs, disentangles sequence polymorphisms in single experiments and will ultimately democratize RNA-structure analysis.
Figures
References
-
- Sharp PA. The centrality of RNA. Cell. 2009;136:577–580. - PubMed
Methods References
-
- Patterson JT, Nickens DG, Burke DH. HIV-1 reverse transcriptase pausing at bulky 2’adducts is relieved by deletion of the RNase H domain. RNA Biol. 2006;3:163. - PubMed
-
- Matathias A, Fox D, Crouse J. SuperScript II RNase H reverse transcriptase. 18064-3. Focus On, Life Technologies; 1999.
-
- Beckman RA, Mildvan AS, Loeb LA. On the fidelity of DNA replication: manganese mutagenesis in vitro. Biochemistry. 1985;24:5810–5817. - PubMed
-
- Wilkinson KA, Merino EJ, Weeks KM. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc. 2006;1:1610–1616. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
