

Annexin V incorporated into influenza virus particles inhibits gamma interferon signaling and promotes viral replication
- PMID: 25031344
- PMCID: PMC4178827
- DOI: 10.1128/JVI.01405-14
Annexin V incorporated into influenza virus particles inhibits gamma interferon signaling and promotes viral replication
Abstract
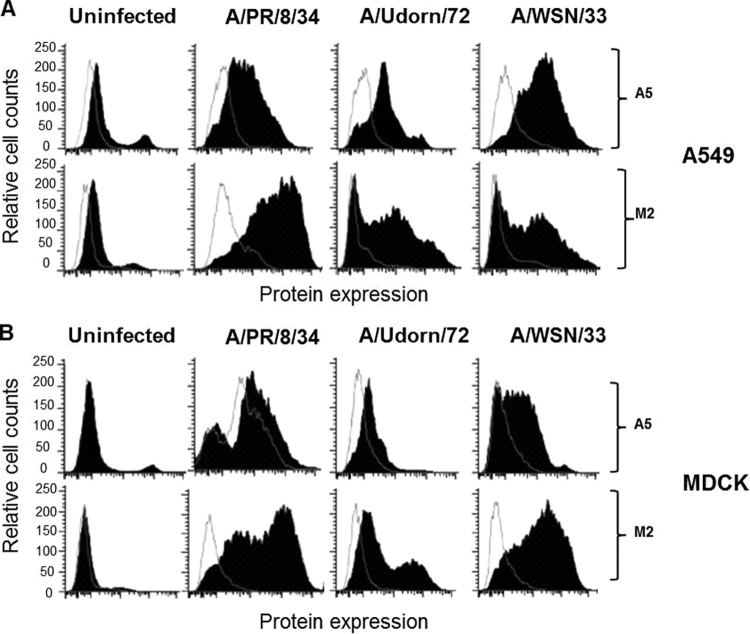
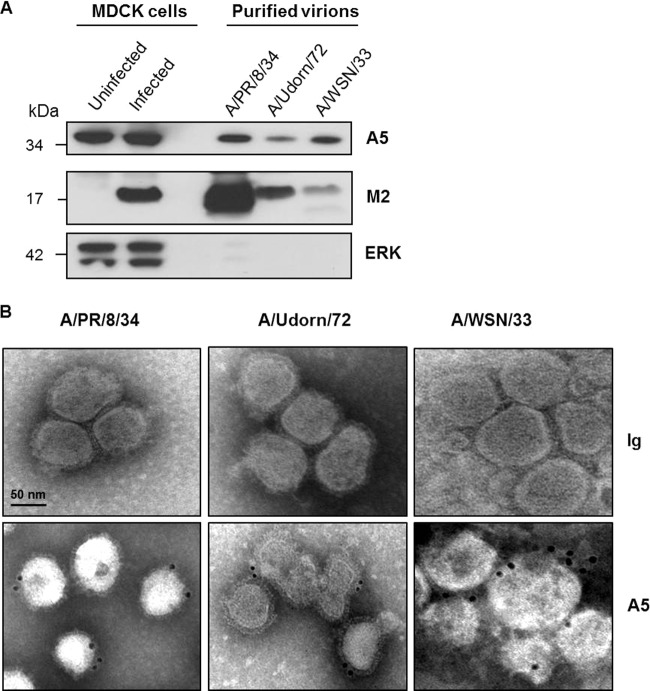
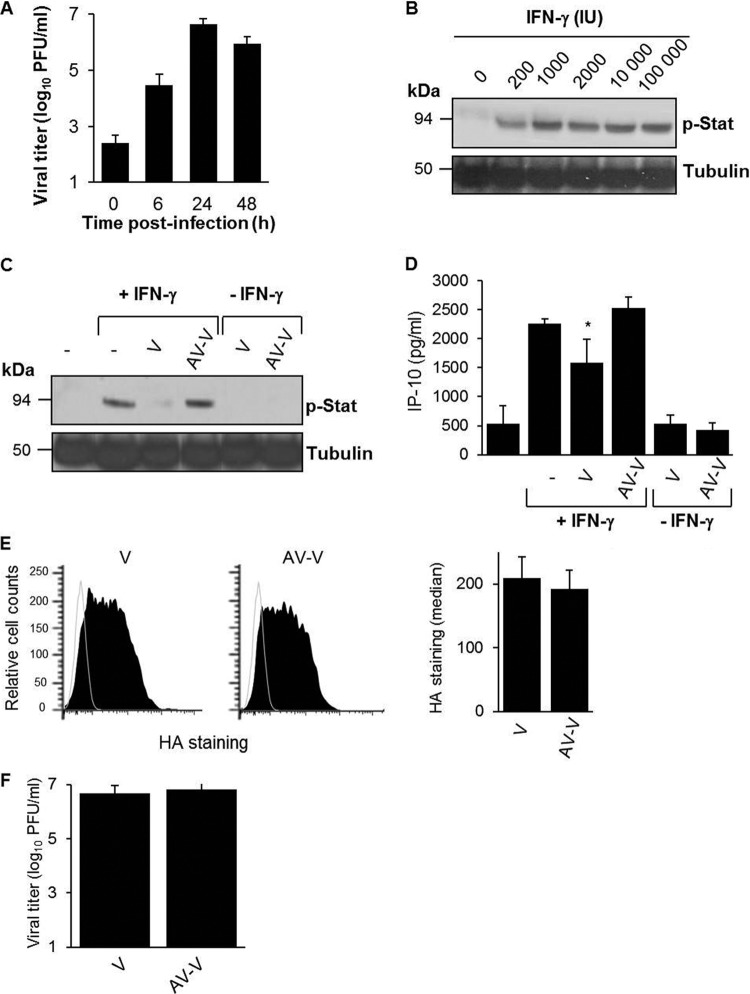
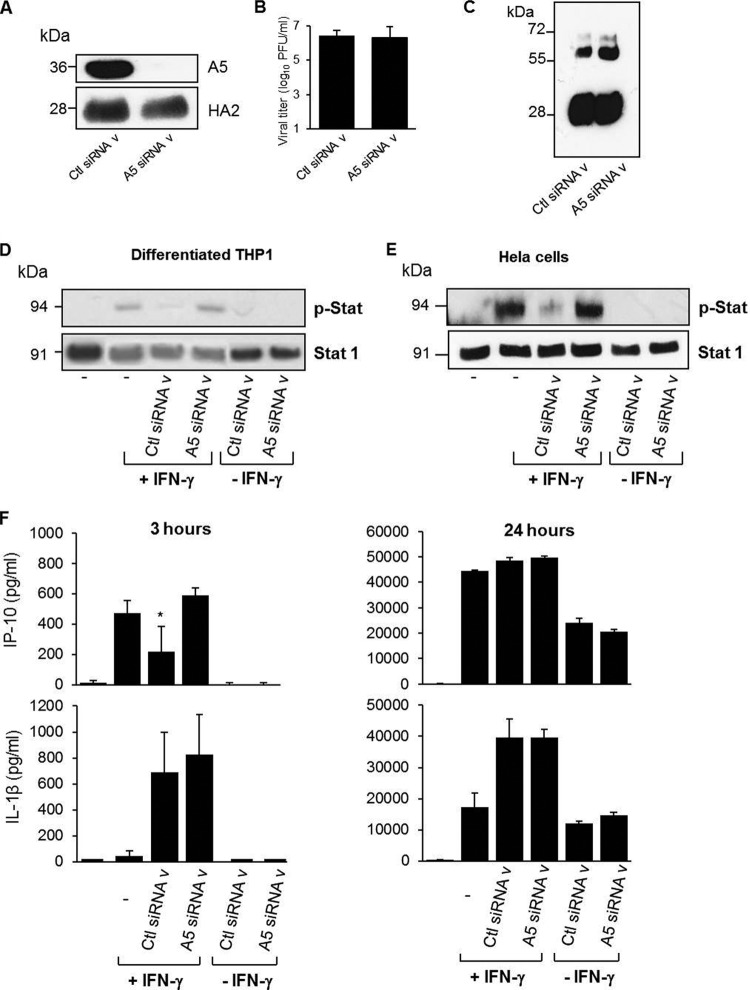
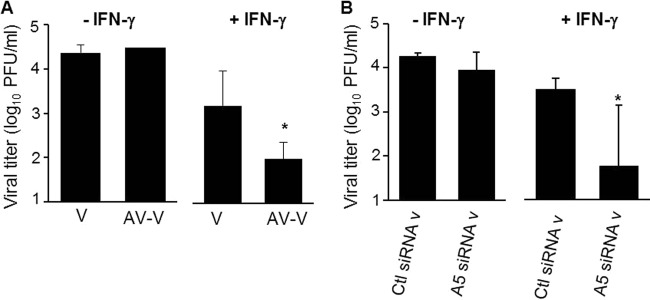
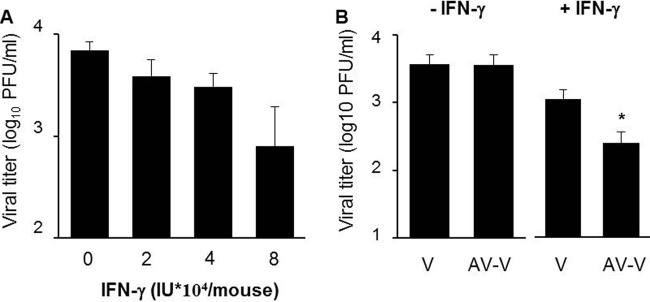
During the budding process, influenza A viruses (IAVs) incorporate multiple host cell membrane proteins. However, for most of them, their significance in viral morphogenesis and infectivity remains unknown. We demonstrate here that the expression of annexin V (A5) is upregulated at the cell surface upon IAV infection and that a substantial proportion of the protein is present in lipid rafts, the site of virus budding. Western blotting and immunogold analysis of highly purified IAV particles showed the presence of A5 in the virion. Significantly, gamma interferon (IFN-γ)-induced Stat phosphorylation and IFN-γ-induced 10-kDa protein (IP-10) production in macrophage-derived THP-1 cells was inhibited by purified IAV particles. Disruption of the IFN-γ signaling pathway was A5 dependent since downregulation of its expression or its blockage reversed the inhibition and resulted in decreased viral replication in vitro. The functional significance of these results was also observed in vivo. Thus, IAVs can subvert the IFN-γ antiviral immune response by incorporating A5 into their envelope during the budding process.
Importance: Many enveloped viruses, including influenza A viruses, bud from the plasma membrane of their host cells and incorporate cellular surface proteins into viral particles. However, for the vast majority of these proteins, only the observation of their incorporation has been reported. We demonstrate here that the host protein annexin V is specifically incorporated into influenza virus particles during the budding process. Importantly, we showed that packaged annexin V counteracted the antiviral activity of gamma interferon in vitro and in vivo. Thus, these results showed that annexin V incorporated in the viral envelope of influenza viruses allow viral escape from immune surveillance. Understanding the role of host incorporated protein into virions may reveal how enveloped RNA viruses hijack the host cell machinery for their own purposes.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
