

NCI-60 whole exome sequencing and pharmacological CellMiner analyses
- PMID: 25032700
- PMCID: PMC4102467
- DOI: 10.1371/journal.pone.0101670
NCI-60 whole exome sequencing and pharmacological CellMiner analyses
Abstract
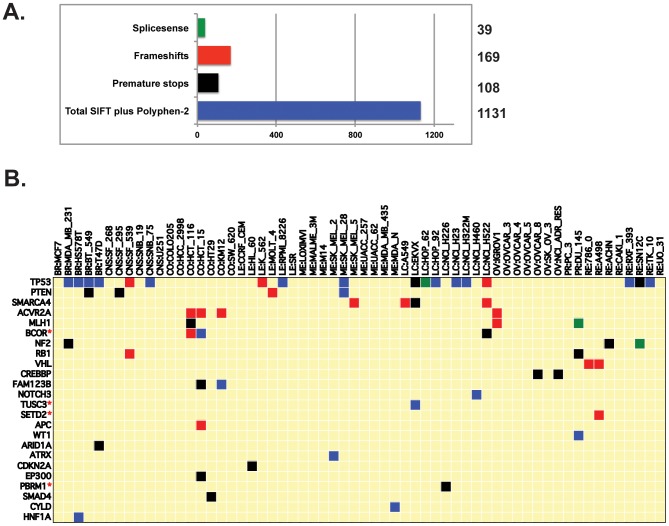
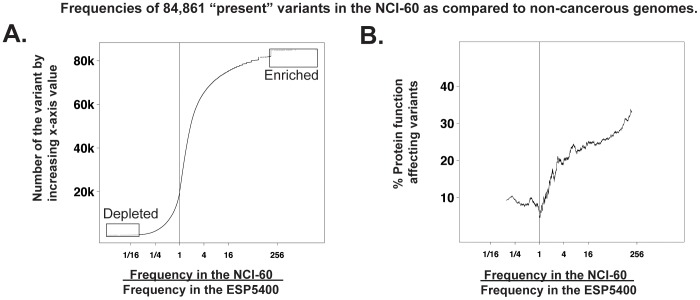
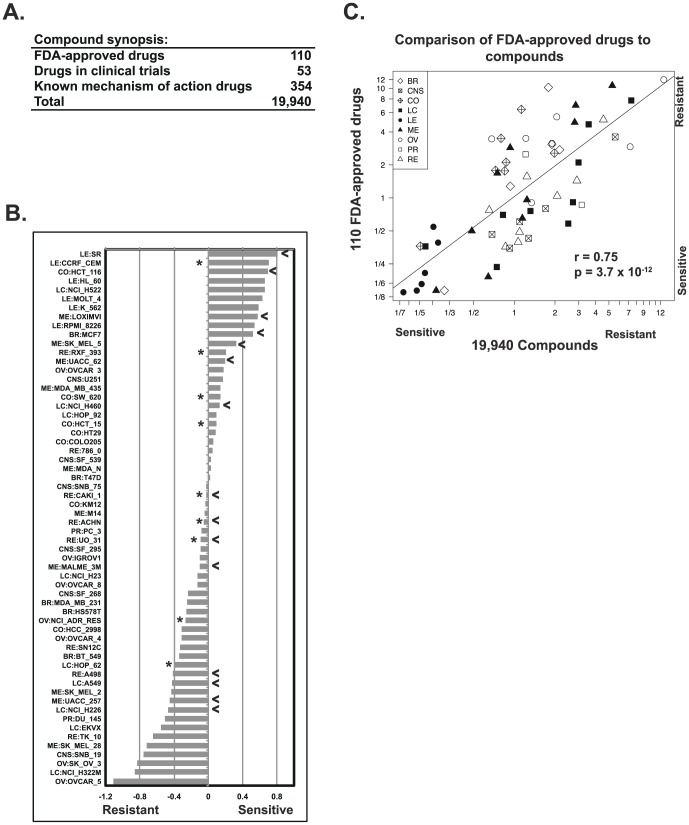
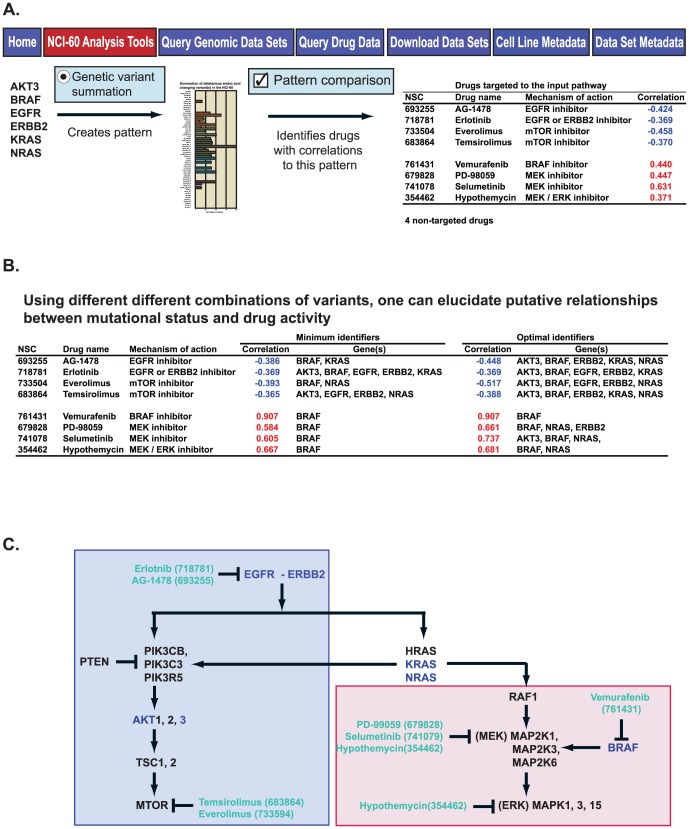
Exome sequencing provides unprecedented insights into cancer biology and pharmacological response. Here we assess these two parameters for the NCI-60, which is among the richest genomic and pharmacological publicly available cancer cell line databases. Homozygous genetic variants that putatively affect protein function were identified in 1,199 genes (approximately 6% of all genes). Variants that are either enriched or depleted compared to non-cancerous genomes, and thus may be influential in cancer progression and differential drug response were identified for 2,546 genes. Potential gene knockouts are made available. Assessment of cell line response to 19,940 compounds, including 110 FDA-approved drugs, reveals ≈80-fold range in resistance versus sensitivity response across cell lines. 103,422 gene variants were significantly correlated with at least one compound (at p<0.0002). These include genes of known pharmacological importance such as IGF1R, BRAF, RAD52, MTOR, STAT2 and TSC2 as well as a large number of candidate genes such as NOM1, TLL2, and XDH. We introduce two new web-based CellMiner applications that enable exploration of variant-to-compound relationships for a broad range of researchers, especially those without bioinformatics support. The first tool, "Genetic variant versus drug visualization", provides a visualization of significant correlations between drug activity-gene variant combinations. Examples are given for the known vemurafenib-BRAF, and novel ifosfamide-RAD52 pairings. The second, "Genetic variant summation" allows an assessment of cumulative genetic variations for up to 150 combined genes together; and is designed to identify the variant burden for molecular pathways or functional grouping of genes. An example of its use is provided for the EGFR-ERBB2 pathway gene variant data and the identification of correlated EGFR, ERBB2, MTOR, BRAF, MEK and ERK inhibitors. The new tools are implemented as an updated web-based CellMiner version, for which the present publication serves as a compendium.
Conflict of interest statement
Figures



References
-
- Doherty D, Bamshad MJ (2012) Exome sequencing to find rare variants causing neurologic diseases. Neurology. - PubMed
-
- Moldovan F, Patten SA, Fendri K, Girard S, Zaouter C, et al. (2012) Exome sequencing identifies novel candidate mutations in idiopathic. Stud Health Technol Inform 176: 453.
-
- Choi BO, Koo SK, Park MH, Rhee H, Yang SJ, et al... (2012) Exome sequencing is an efficient tool for genetic screening of Charcot-Marie-Tooth Disease. Hum Mutat. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
