

Endothelial arginine resynthesis contributes to the maintenance of vasomotor function in male diabetic mice
- PMID: 25033204
- PMCID: PMC4102520
- DOI: 10.1371/journal.pone.0102264
Endothelial arginine resynthesis contributes to the maintenance of vasomotor function in male diabetic mice
Abstract
Aim: Argininosuccinate synthetase (ASS) is essential for recycling L-citrulline, the by-product of NO synthase (NOS), to the NOS substrate L-arginine. Here, we assessed whether disturbed arginine resynthesis modulates endothelium-dependent vasodilatation in normal and diabetic male mice.
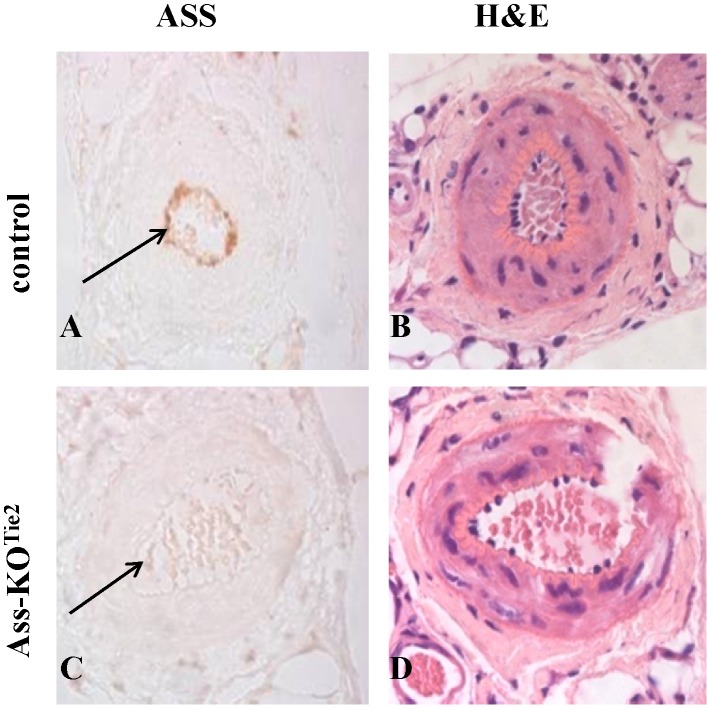
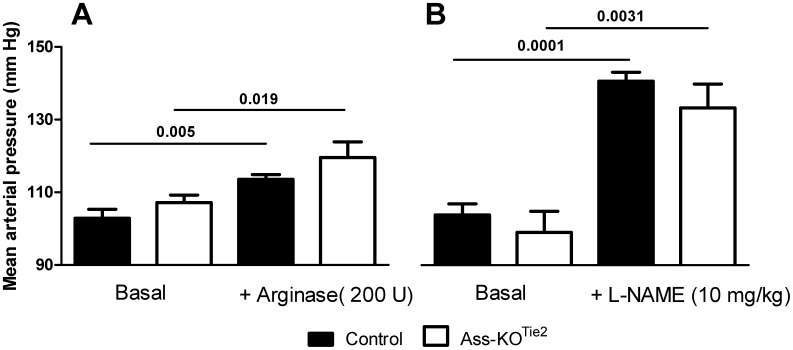
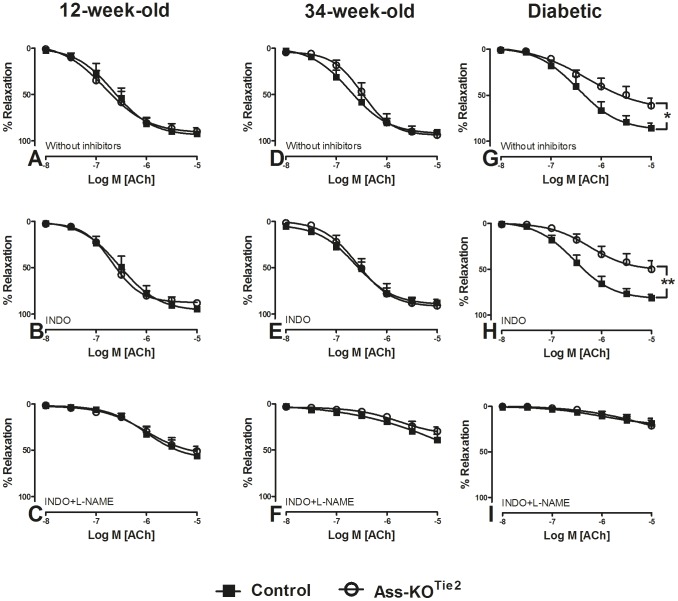
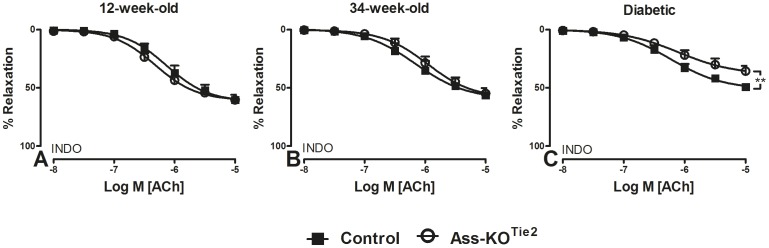
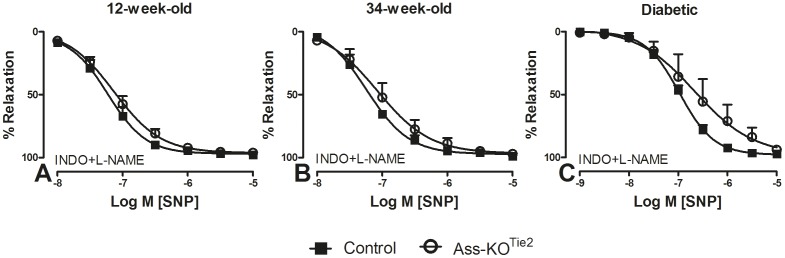
Methods and results: Endothelium-selective Ass-deficient mice (Assfl/fl/Tie2Cretg/- = Ass-KOTie2) were generated by crossing Assfl/fl mice ( = control) with Tie2Cre mice. Gene ablation in endothelial cells was confirmed by immunohistochemistry. Blood pressure (MAP) was recorded in 34-week-old male mice. Vasomotor responses were studied in isolated saphenous arteries of 12- and 34-week-old Ass-KOTie2 and control animals. At the age of 10 weeks, diabetes was induced in control and Ass-KOTie2 mice by streptozotocin injections. Vasomotor responses of diabetic animals were studied 10 weeks later. MAP was similar in control and Ass-KOTie2 mice. Depletion of circulating L-arginine by arginase 1 infusion or inhibition of NOS activity with L-NAME resulted in an increased MAP (10 and 30 mmHg, respectively) in control and Ass-KOTie2 mice. Optimal arterial diameter, contractile responses to phenylephrine, and relaxing responses to acetylcholine and sodium nitroprusside were similar in healthy control and Ass-KOTie2 mice. However, in diabetic Ass-KOTie2 mice, relaxation responses to acetylcholine and endothelium-derived NO (EDNO) were significantly reduced when compared to diabetic control mice.
Conclusions: Absence of endothelial citrulline recycling to arginine did not affect blood pressure and systemic arterial vasomotor responses in healthy mice. EDNO-mediated vasodilatation was significantly more impaired in diabetic Ass-KOTie2 than in control mice demonstrating that endothelial arginine recycling becomes a limiting endothelial function in diabetes.
Conflict of interest statement
Figures
References
-
- Radomski MW, Palmer RM, Moncada S (1987) Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet 2: 1057–1058. - PubMed
-
- Hogg N, Kalyanaraman B, Joseph J, Struck A, Parthasarathy S (1993) Inhibition of low-density lipoprotein oxidation by nitric oxide. Potential role in atherogenesis. FEBS Lett 334: 170–174. - PubMed
-
- Taddei S, Virdis A, Mattei P, Ghiadoni L, Sudano I, et al. (1996) Defective L-arginine-nitric oxide pathway in offspring of essential hypertensive patients. Circulation 94: 1298–1303. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
