

Enhanced regulatory sequence prediction using gapped k-mer features
- PMID: 25033408
- PMCID: PMC4102394
- DOI: 10.1371/journal.pcbi.1003711
Enhanced regulatory sequence prediction using gapped k-mer features
Erratum in
- PLoS Comput Biol. 2014 Dec;10(12):e1004035
Abstract
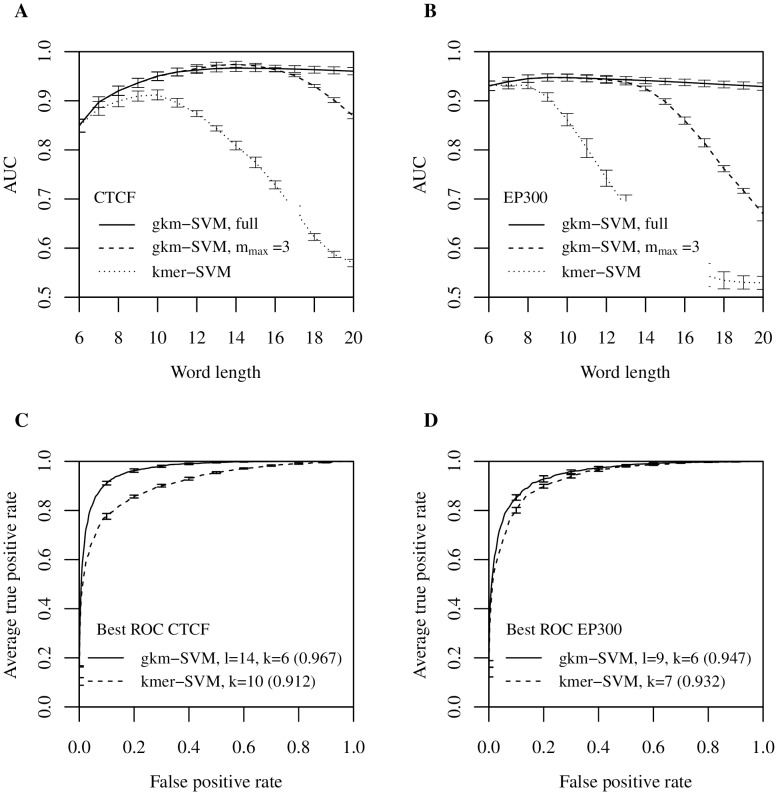
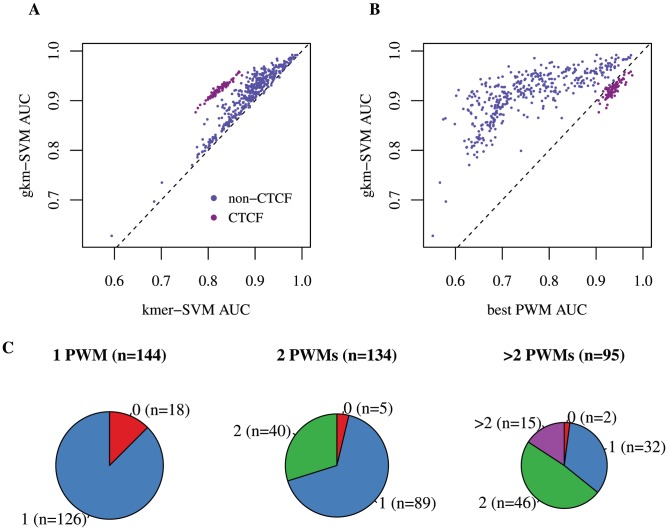
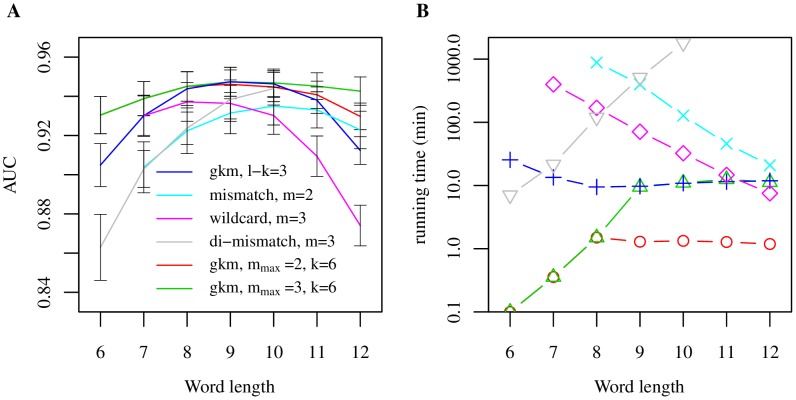
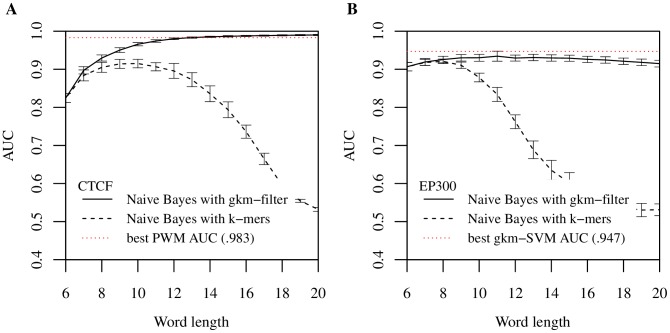
Oligomers of length k, or k-mers, are convenient and widely used features for modeling the properties and functions of DNA and protein sequences. However, k-mers suffer from the inherent limitation that if the parameter k is increased to resolve longer features, the probability of observing any specific k-mer becomes very small, and k-mer counts approach a binary variable, with most k-mers absent and a few present once. Thus, any statistical learning approach using k-mers as features becomes susceptible to noisy training set k-mer frequencies once k becomes large. To address this problem, we introduce alternative feature sets using gapped k-mers, a new classifier, gkm-SVM, and a general method for robust estimation of k-mer frequencies. To make the method applicable to large-scale genome wide applications, we develop an efficient tree data structure for computing the kernel matrix. We show that compared to our original kmer-SVM and alternative approaches, our gkm-SVM predicts functional genomic regulatory elements and tissue specific enhancers with significantly improved accuracy, increasing the precision by up to a factor of two. We then show that gkm-SVM consistently outperforms kmer-SVM on human ENCODE ChIP-seq datasets, and further demonstrate the general utility of our method using a Naïve-Bayes classifier. Although developed for regulatory sequence analysis, these methods can be applied to any sequence classification problem.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
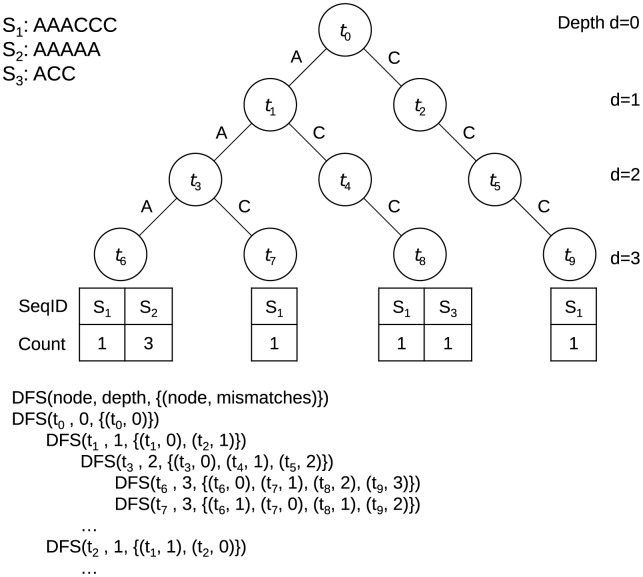
References
-
- Beer MA, Tavazoie S (2004) Predicting Gene Expression from Sequence. Cell 117: 185–198. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
