

High-throughput functional testing of ENCODE segmentation predictions
- PMID: 25035418
- PMCID: PMC4199366
- DOI: 10.1101/gr.173518.114
High-throughput functional testing of ENCODE segmentation predictions
Abstract
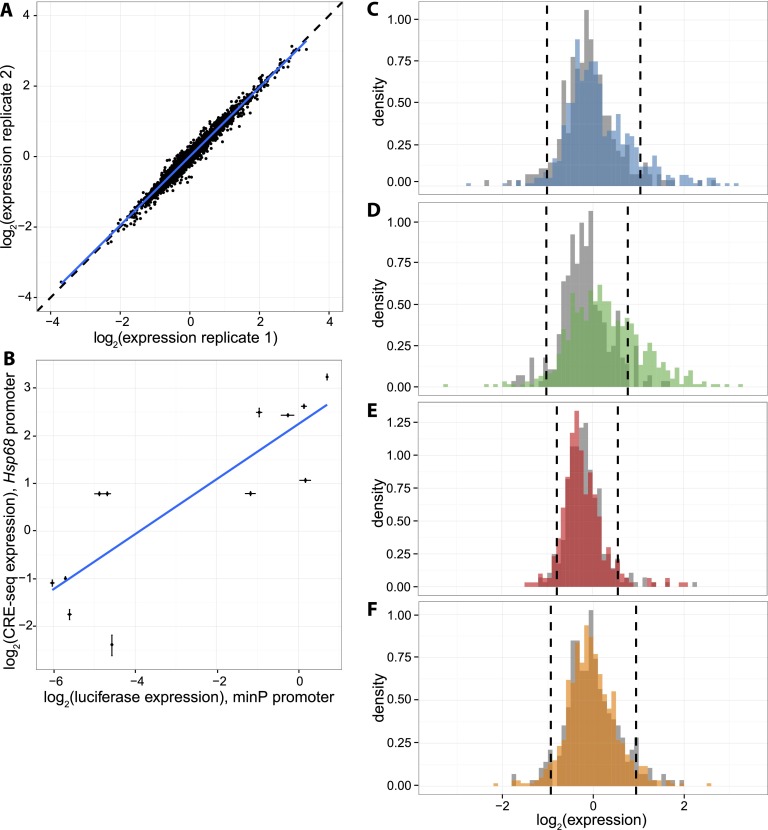
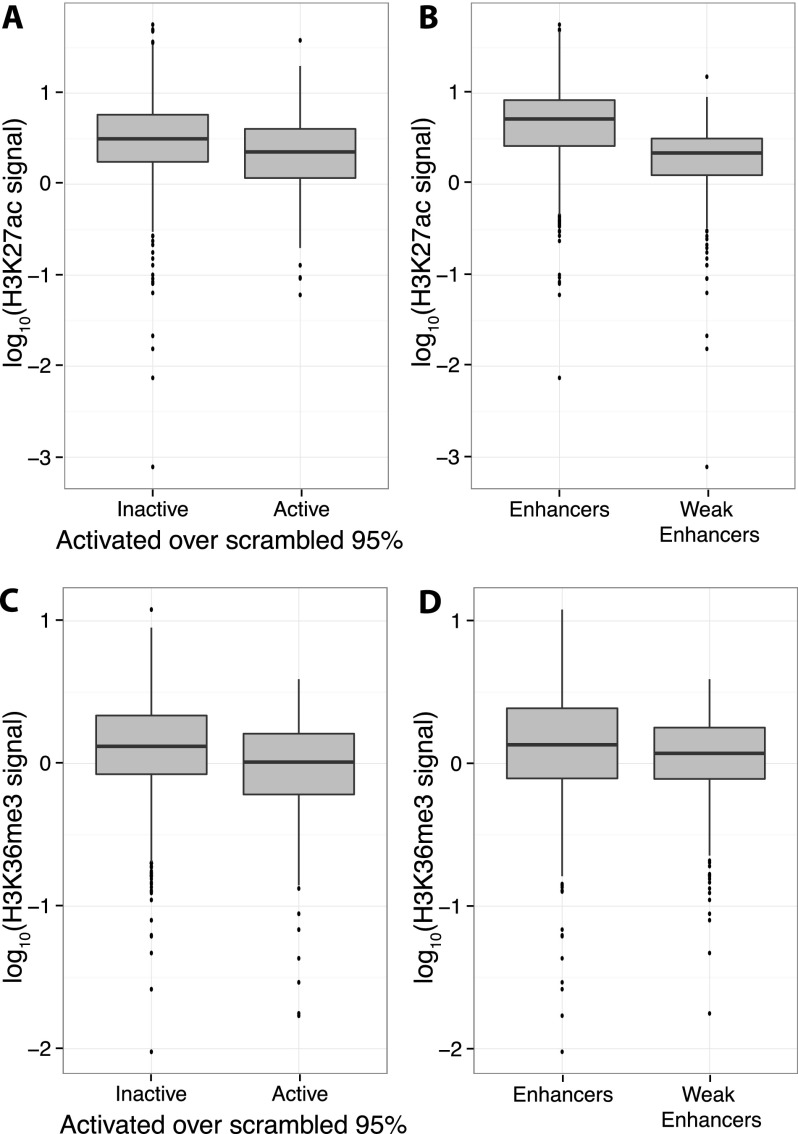
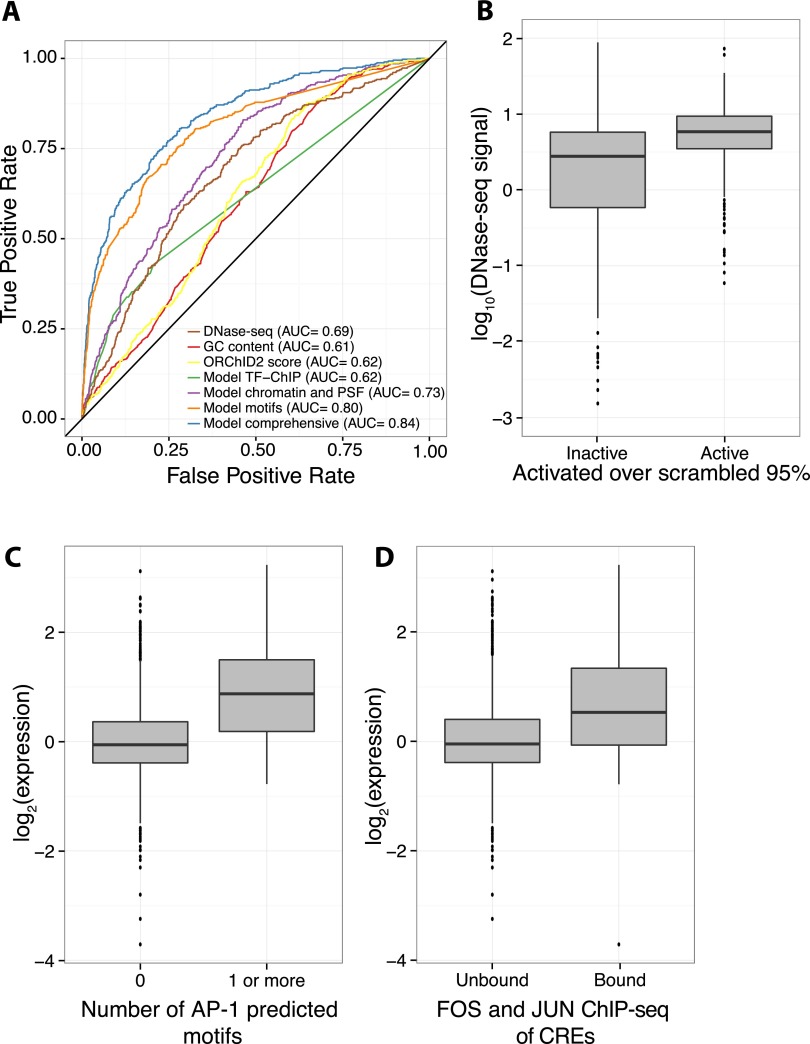
The histone modification state of genomic regions is hypothesized to reflect the regulatory activity of the underlying genomic DNA. Based on this hypothesis, the ENCODE Project Consortium measured the status of multiple histone modifications across the genome in several cell types and used these data to segment the genome into regions with different predicted regulatory activities. We measured the cis-regulatory activity of more than 2000 of these predictions in the K562 leukemia cell line. We tested genomic segments predicted to be Enhancers, Weak Enhancers, or Repressed elements in K562 cells, along with other sequences predicted to be Enhancers specific to the H1 human embryonic stem cell line (H1-hESC). Both Enhancer and Weak Enhancer sequences in K562 cells were more active than negative controls, although surprisingly, Weak Enhancer segmentations drove expression higher than did Enhancer segmentations. Lower levels of the covalent histone modifications H3K36me3 and H3K27ac, thought to mark active enhancers and transcribed gene bodies, associate with higher expression and partly explain the higher activity of Weak Enhancers over Enhancer predictions. While DNase I hypersensitivity (HS) is a good predictor of active sequences in our assay, transcription factor (TF) binding models need to be included in order to accurately identify highly expressed sequences. Overall, our results show that a significant fraction (-26%) of the ENCODE enhancer predictions have regulatory activity, suggesting that histone modification states can reflect the cis-regulatory activity of sequences in the genome, but that specific sequence preferences, such as TF-binding sites, are the causal determinants of cis-regulatory activity.
© 2014 Kwasnieski et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Akaike H. 1974. A new look at the statistical model identification. IEEE Trans Automat Contr 19: 716–723
-
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. 2007. High-resolution profiling of histone methylations in the human genome. Cell 129: 823–837 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous