

Mitochondrial EFTs defects in juvenile-onset Leigh disease, ataxia, neuropathy, and optic atrophy
- PMID: 25037205
- PMCID: PMC4150129
- DOI: 10.1212/WNL.0000000000000716
Mitochondrial EFTs defects in juvenile-onset Leigh disease, ataxia, neuropathy, and optic atrophy
Abstract
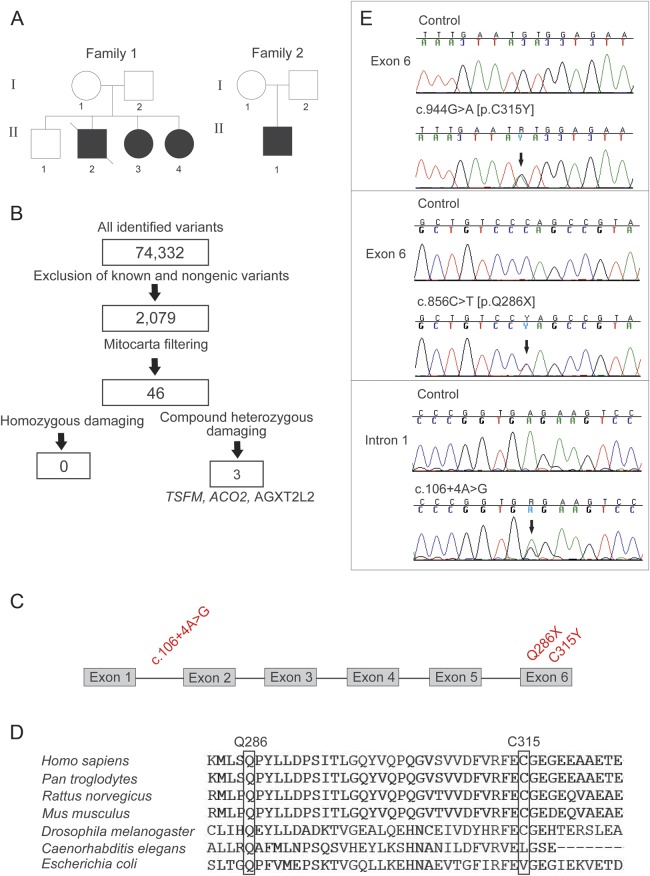
Objective: We report novel defects of mitochondrial translation elongation factor Ts (EFTs), with high carrier frequency in Finland and expand the manifestations of this disease group from infantile cardiomyopathy to juvenile neuropathy/encephalopathy disorders.
Methods: DNA analysis, whole-exome analysis, protein biochemistry, and protein modeling.
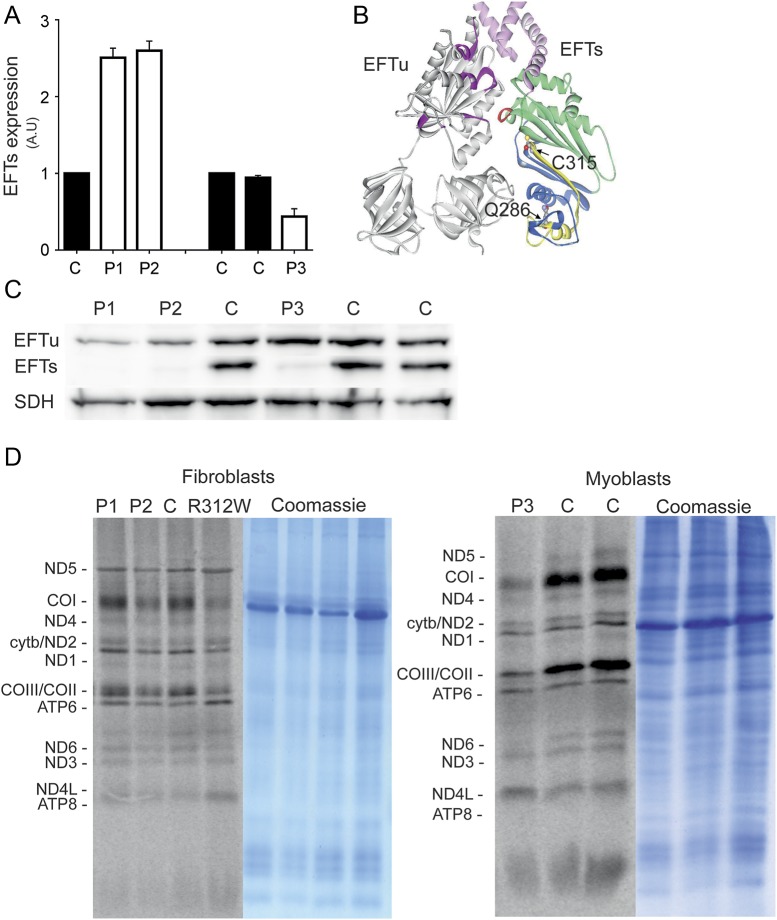
Results: We used whole-exome sequencing to find the genetic cause of infantile-onset mitochondrial cardiomyopathy, progressing to juvenile-onset Leigh syndrome, neuropathy, and optic atrophy in 2 siblings. We found novel compound heterozygous mutations, c.944G>A [p.C315Y] and c.856C>T [p.Q286X], in the TSFM gene encoding mitochondrial EFTs. The same p.Q286X variant was found as compound heterozygous with a splice site change in a patient from a second family, with juvenile-onset optic atrophy, peripheral neuropathy, and ataxia. Our molecular modeling predicted the coding-region mutations to cause protein instability, which was experimentally confirmed in cultured patient cells, with mitochondrial translation defect and lacking EFTs. Only a single TSFM mutation has been previously described in different populations, leading to an infantile fatal multisystem disorder with cardiomyopathy. Sequence data from 35,000 Finnish population controls indicated that the heterozygous carrier frequency of p.Q286X change was exceptionally high in Finland, 1:80, but no homozygotes were found in the population, in our mitochondrial disease patient collection, or in an intrauterine fetal death material, suggesting early developmental lethality of the homozygotes.
Conclusions: We show that in addition to early-onset cardiomyopathy, TSFM mutations should be considered in childhood and juvenile encephalopathies with optic and/or peripheral neuropathy, ataxia, or Leigh disease.
© 2014 American Academy of Neurology.
Figures
References
-
- Ylikallio E, Suomalainen A. Mechanisms of mitochondrial diseases. Ann Med 2012;44:41–59 - PubMed
-
- Antonicka H, Sasarman F, Kennaway NG, Shoubridge EA. The molecular basis for tissue specificity of the oxidative phosphorylation deficiencies in patients with mutations in the mitochondrial translation factor EFG1. Hum Mol Genet 2006;15:1835–1846 - PubMed
-
- Coenen MJ, Antonicka H, Ugalde C, et al. Mutant mitochondrial elongation factor G1 and combined oxidative phosphorylation deficiency. N Engl J Med 2004;351:2080–2086 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources