

How proteins bind macrocycles
- PMID: 25038790
- PMCID: PMC4417626
- DOI: 10.1038/nchembio.1584
How proteins bind macrocycles
Abstract
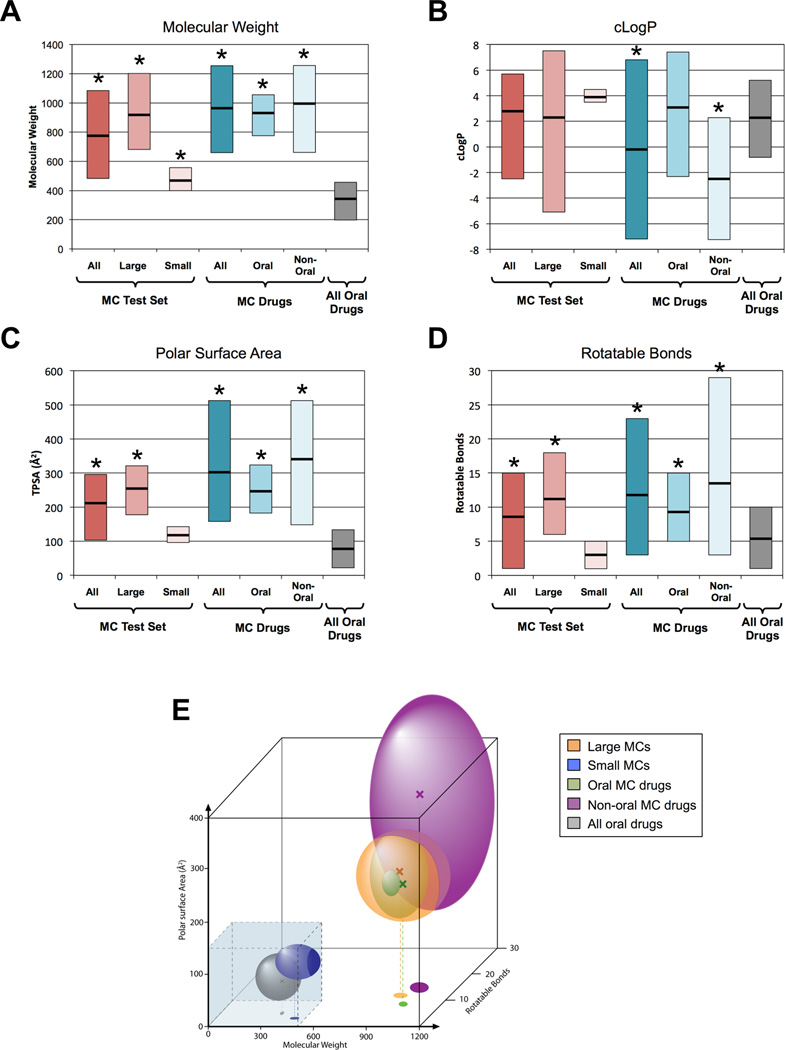
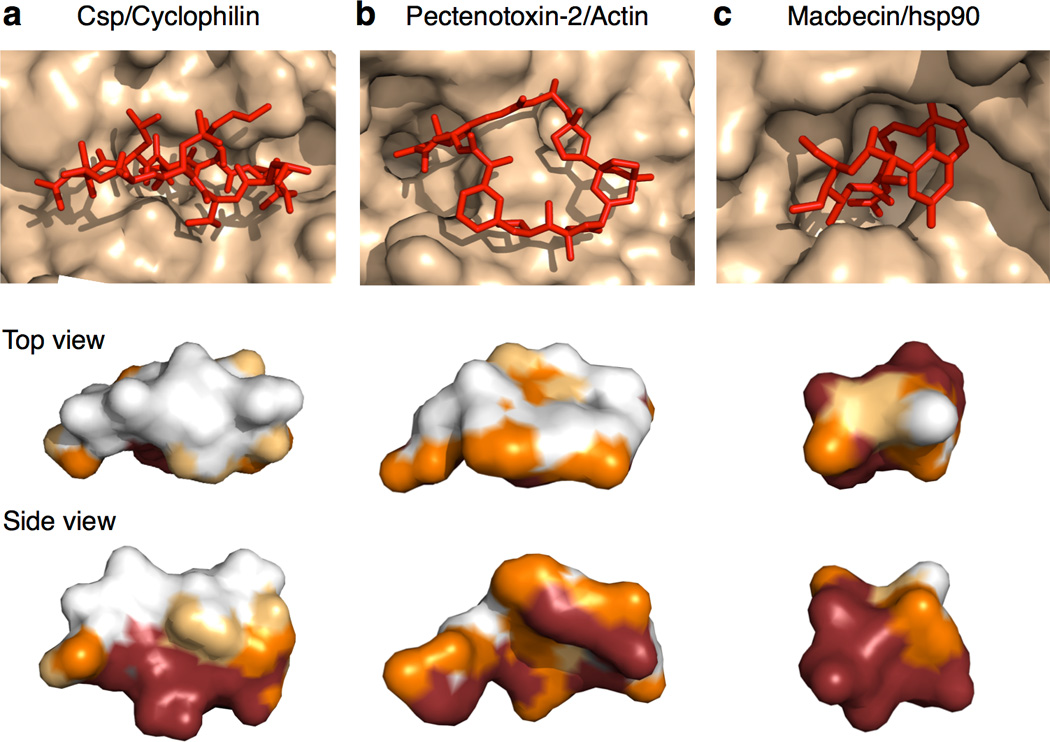
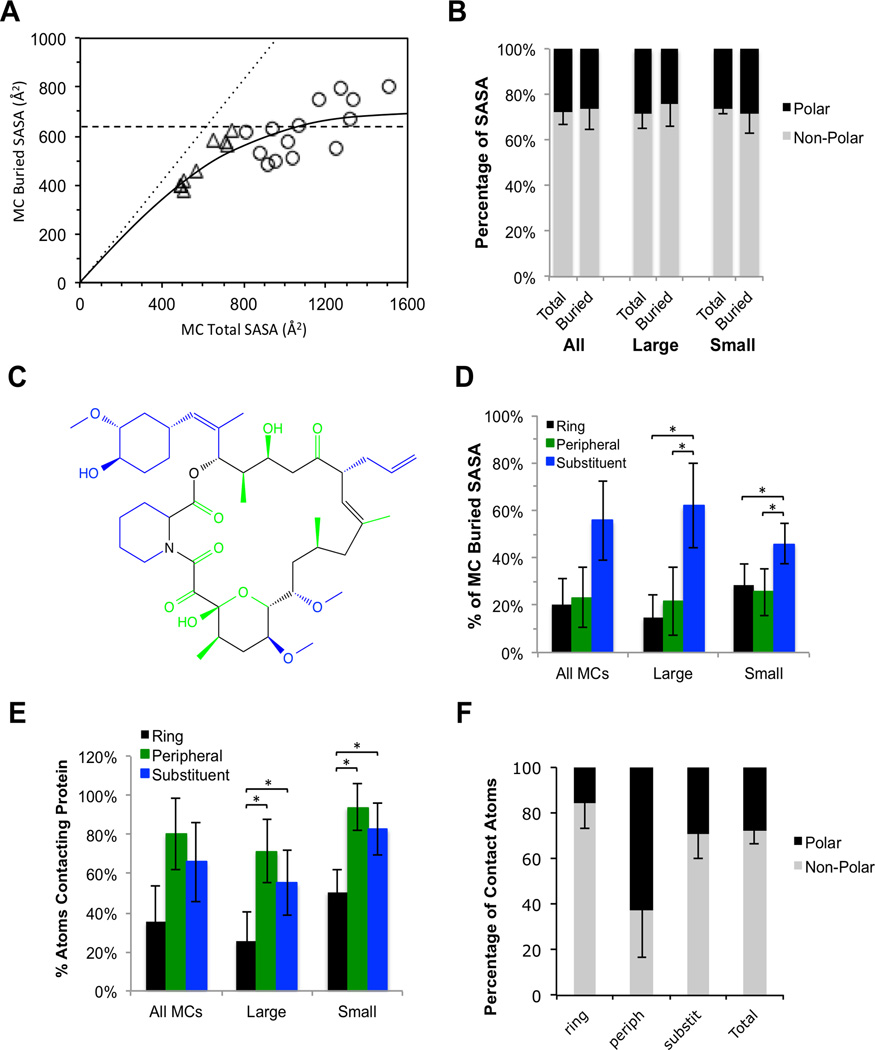
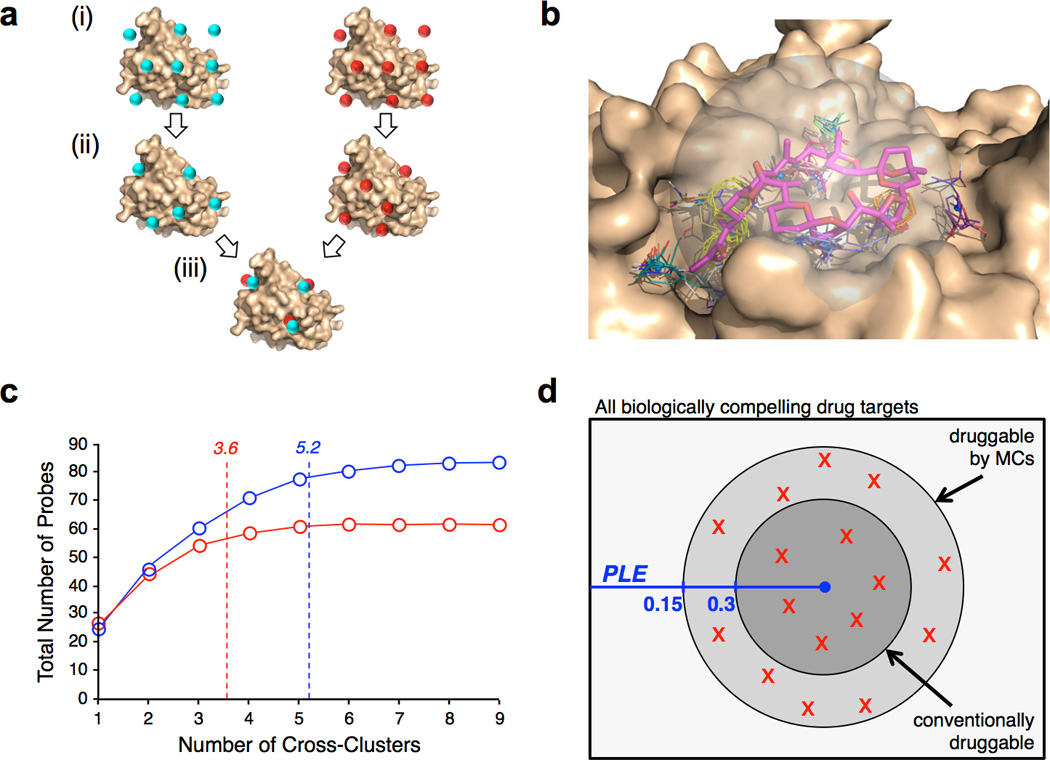
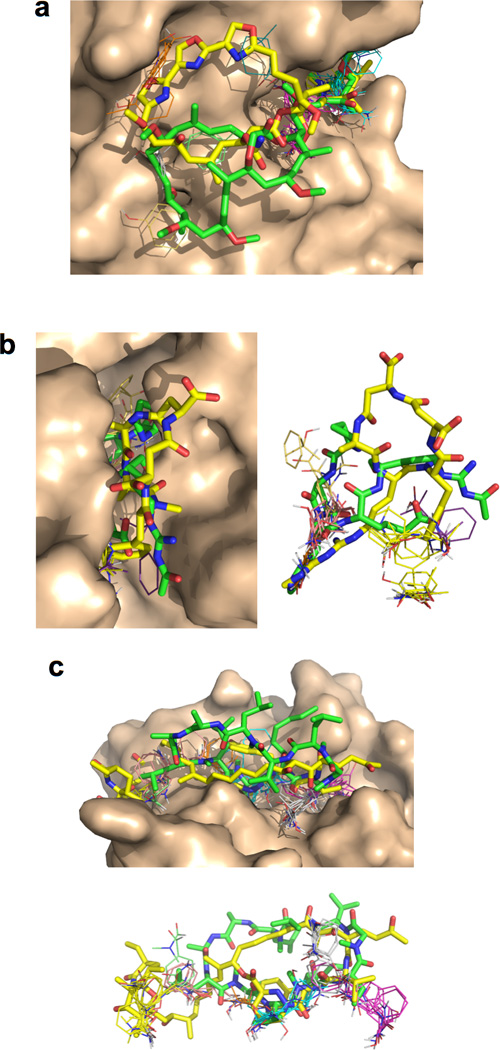
The potential utility of synthetic macrocycles (MCs) as drugs, particularly against low-druggability targets such as protein-protein interactions, has been widely discussed. There is little information, however, to guide the design of MCs for good target protein-binding activity or bioavailability. To address this knowledge gap, we analyze the binding modes of a representative set of MC-protein complexes. The results, combined with consideration of the physicochemical properties of approved macrocyclic drugs, allow us to propose specific guidelines for the design of synthetic MC libraries with structural and physicochemical features likely to favor strong binding to protein targets as well as good bioavailability. We additionally provide evidence that large, natural product-derived MCs can bind targets that are not druggable by conventional, drug-like compounds, supporting the notion that natural product-inspired synthetic MCs can expand the number of proteins that are druggable by synthetic small molecules.
Figures
References
-
- Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–730. - PubMed
-
- Berg T. Small-molecule inhibitors of protein-protein interactions. Curr Opin Drug Discov Devel. 2008;11:666–674. - PubMed
-
- Buchwald P. Small-molecule protein-protein interaction inhibitors: therapeutic potential in light of molecular size, chemical space, and ligand binding efficiency considerations. IUBMB life. 2010;62:724–731. - PubMed
-
- Whitty A, Kumaravel G. Between a rock and a hard place? Nat Chem Biol. 2006;2:112–118. - PubMed
-
- Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
