

Mutation of POC1B in a severe syndromic retinal ciliopathy
- PMID: 25044745
- PMCID: PMC4425427
- DOI: 10.1002/humu.22618
Mutation of POC1B in a severe syndromic retinal ciliopathy
Abstract
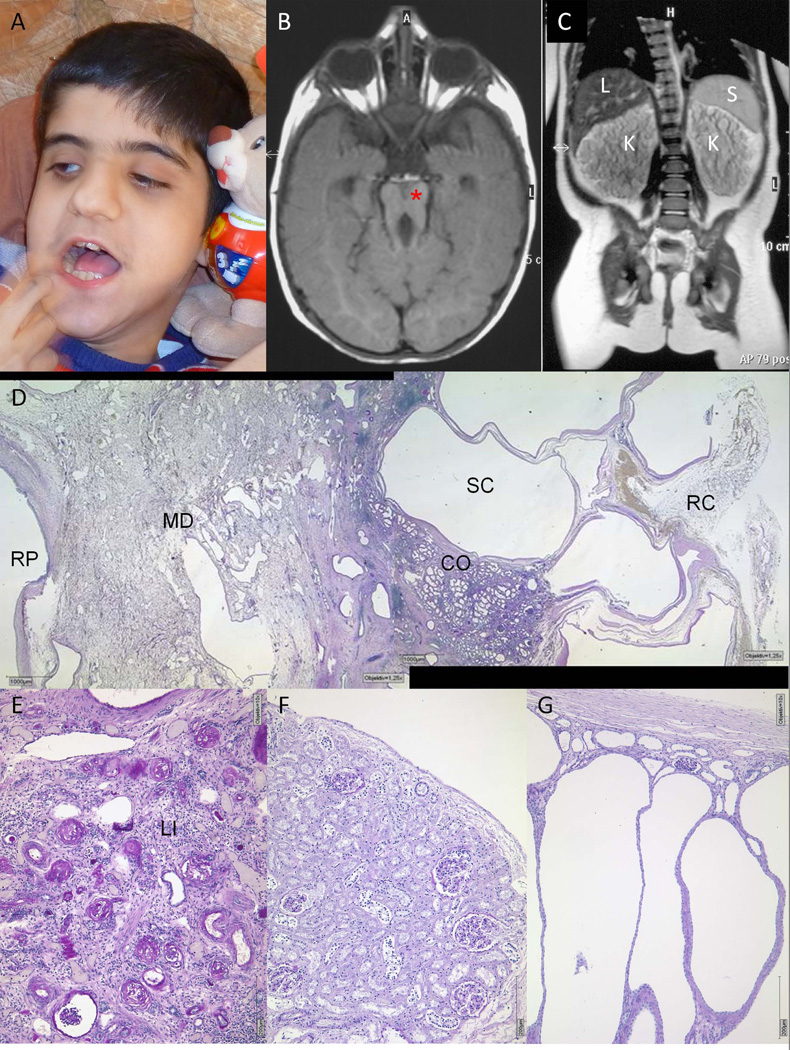
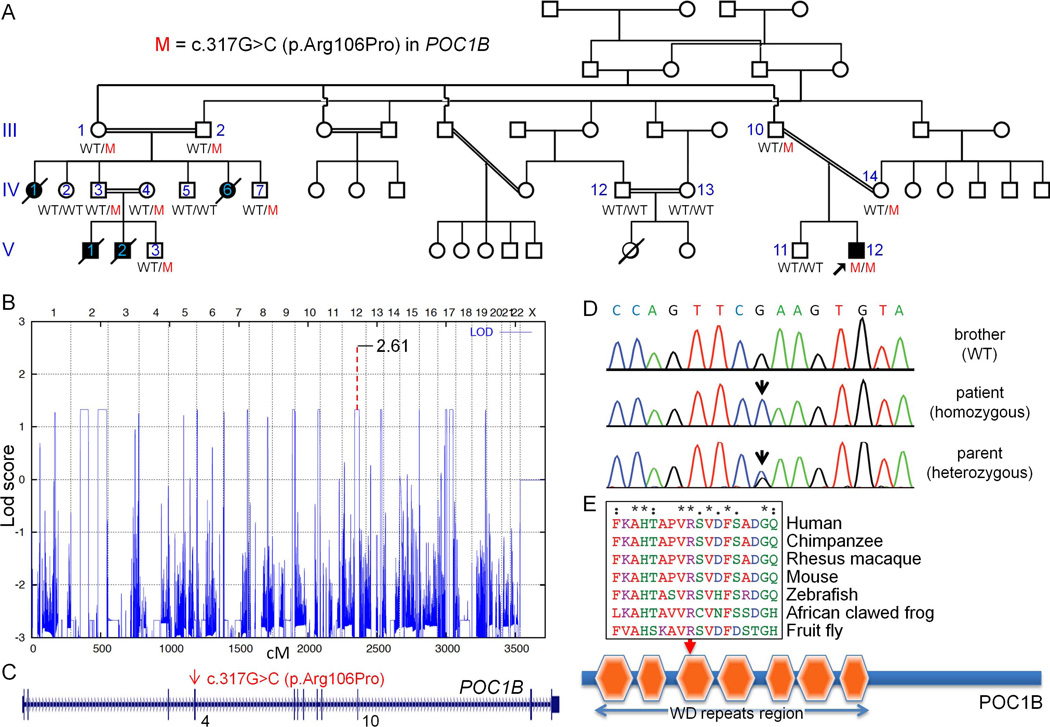
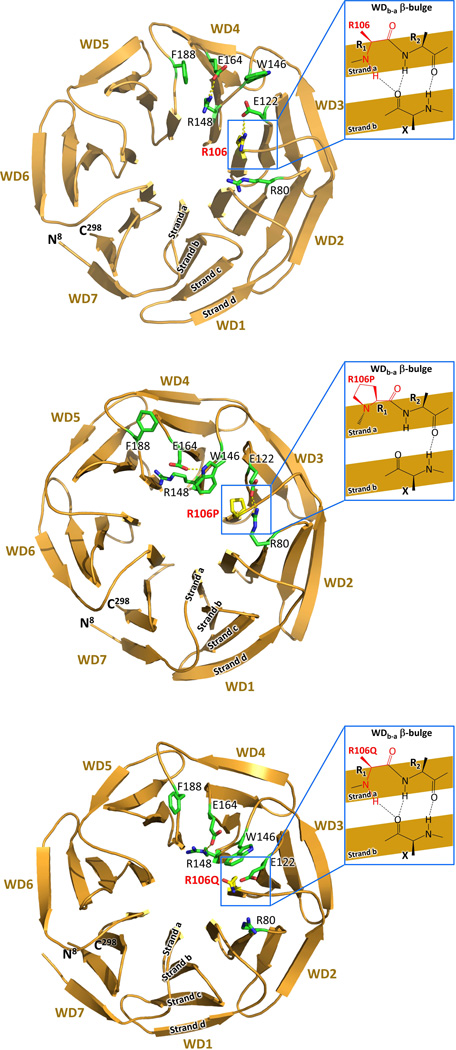
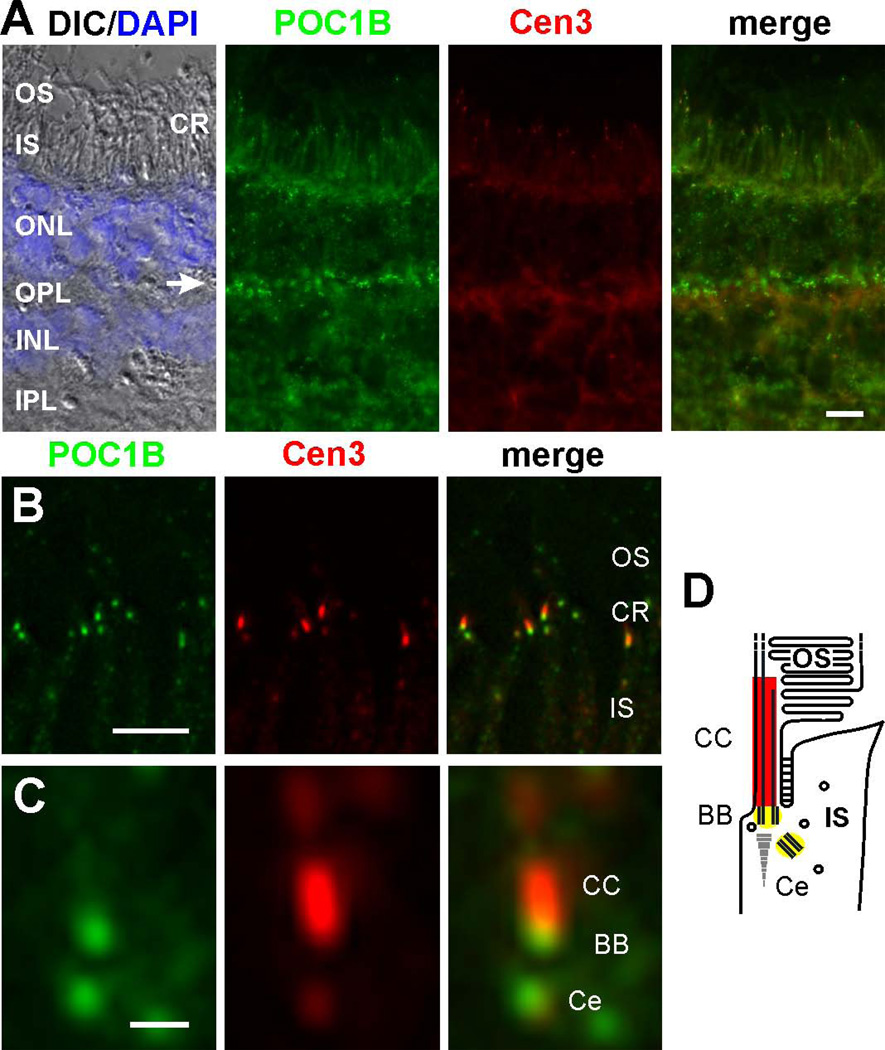
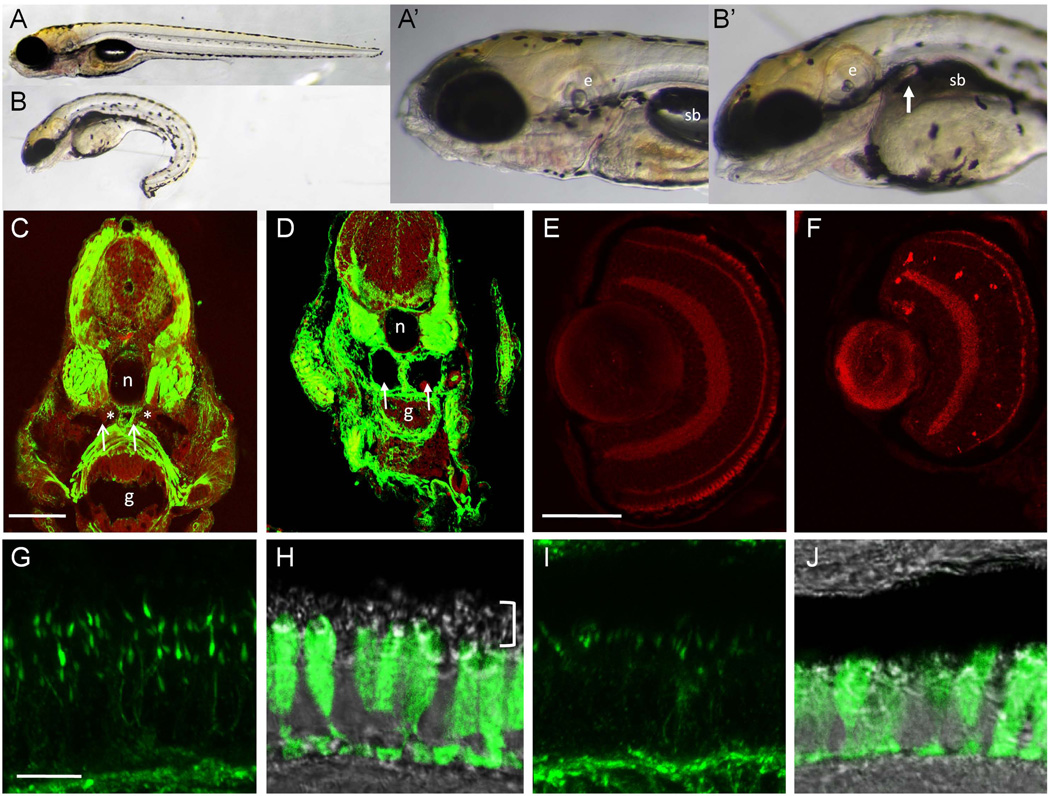
We describe a consanguineous Iraqi family with Leber congenital amaurosis (LCA), Joubert syndrome (JBTS), and polycystic kidney disease (PKD). Targeted next-generation sequencing for excluding mutations in known LCA and JBTS genes, homozygosity mapping, and whole-exome sequencing identified a homozygous missense variant, c.317G>C (p.Arg106Pro), in POC1B, a gene essential for ciliogenesis, basal body, and centrosome integrity. In silico modeling suggested a requirement of p.Arg106 for the formation of the third WD40 repeat and a protein interaction interface. In human and mouse retina, POC1B localized to the basal body and centriole adjacent to the connecting cilium of photoreceptors and in synapses of the outer plexiform layer. Knockdown of Poc1b in zebrafish caused cystic kidneys and retinal degeneration with shortened and reduced photoreceptor connecting cilia, compatible with the human syndromic ciliopathy. A recent study describes homozygosity for p.Arg106ProPOC1B in a family with nonsyndromic cone-rod dystrophy. The phenotype associated with homozygous p.Arg106ProPOC1B may thus be highly variable, analogous to homozygous p.Leu710Ser in WDR19 causing either isolated retinitis pigmentosa or Jeune syndrome. Our study indicates that POC1B is required for retinal integrity, and we propose POC1B mutations as a probable cause for JBTS with severe PKD.
Keywords: Joubert syndrome; LCA; POC1B; ciliopathy; zebrafish.
© 2014 WILEY PERIODICALS, INC.
Conflict of interest statement
T.E., C.B. and H.J.B. are employees of Bioscientia which is part of a publicly traded diagnostic company. The other authors have no competing interests.
Figures
References
-
- Basmanav FB, Oprisoreanu AM, Pasternack SM, Thiele H, Fritz G, Wenzel J, Grosser L, Wehner M, Wolf S, Fagerberg C, Bygum A, Altmuller J, et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomal-dominant Dowling-Degos disease. Am J Hum Genet. 2014;94:135–143. - PMC - PubMed
-
- Boldt K, Mans DA, Won J, van Reeuwijk J, Vogt A, Kinkl N, Letteboer SJ, Hicks WL, Hurd RE, Naggert JK, Texier Y, den Hollander AI, et al. Disruption of intraflagellar protein transport in photoreceptor cilia causes Leber congenital amaurosis in humans and mice. J Clin Invest. 2011;121:2169–2180. - PMC - PubMed
-
- Coussa RG, Otto EA, Gee HY, Arthurs P, Ren H, Lopez I, Keser V, Fu Q, Faingold R, Khan A, Schwartzentruber J, Majewski J, et al. WDR19: an ancient, retrograde, intraflagellar ciliary protein is mutated in autosomal recessive retinitis pigmentosa and in Senior-Loken syndrome. Clin Genet. 2013;84:150–159. - PMC - PubMed
-
- Durlu YK, Koroglu C, Tolun A. Novel Recessive Cone-Rod Dystrophy Caused by POC1B Mutation. JAMA Ophthalmol. 2014 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
