

Whole exome sequencing identifies three novel mutations in ANTXR1 in families with GAPO syndrome
- PMID: 25045128
- PMCID: PMC4332576
- DOI: 10.1002/ajmg.a.36678
Whole exome sequencing identifies three novel mutations in ANTXR1 in families with GAPO syndrome
Abstract
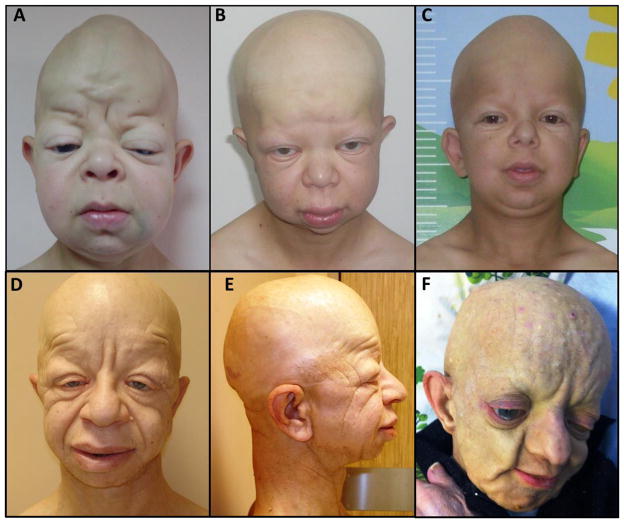
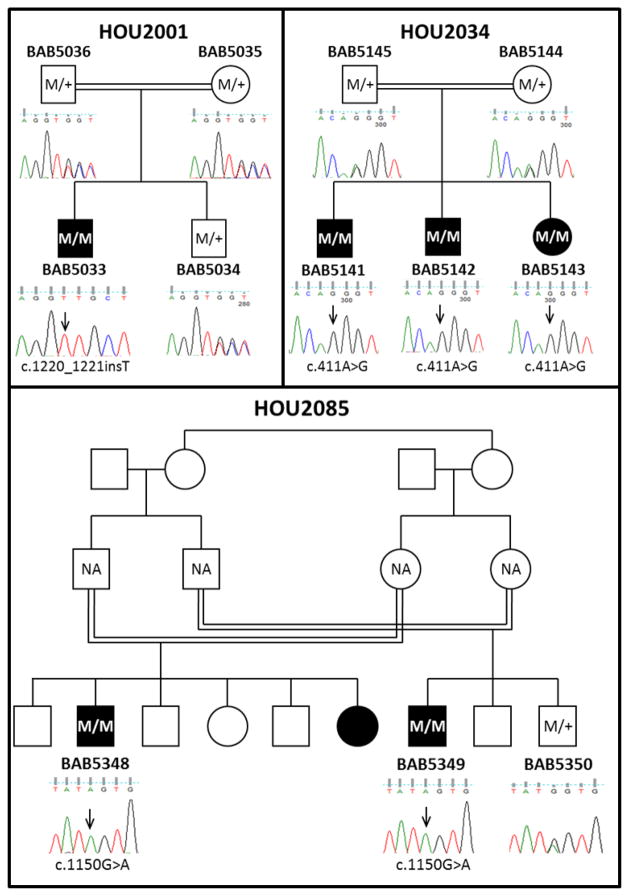
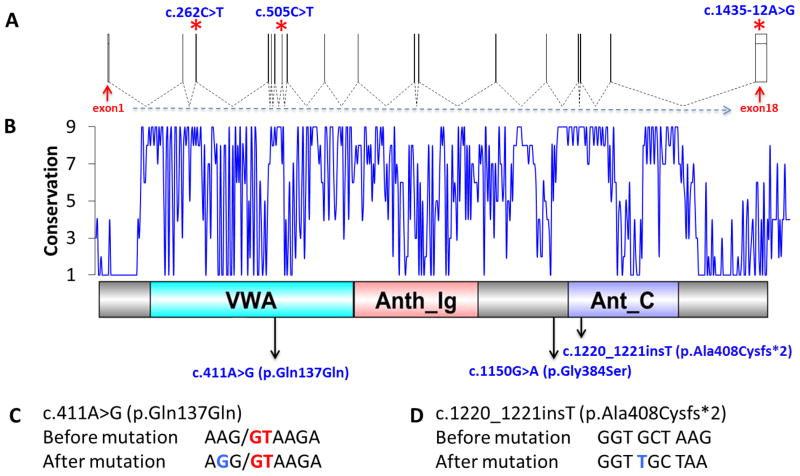
GAPO syndrome (OMIM#230740) is the acronym for growth retardation, alopecia, pseudoanodontia, and optic atrophy. About 35 cases have been reported, making it among one of the rarest recessive conditions. Distinctive craniofacial features including alopecia, rarefaction of eyebrows and eyelashes, frontal bossing, high forehead, mid-facial hypoplasia, hypertelorism, and thickened eyelids and lips make GAPO syndrome a clinically recognizable phenotype. While this genomic study was in progress mutations in ANTXR1 were reported to cause GAPO syndrome. In our study we performed whole exome sequencing (WES) for five affected individuals from three Turkish kindreds segregating the GAPO trait. Exome sequencing analysis identified three novel homozygous mutations including; one frame-shift (c.1220_1221insT; p.Ala408Cysfs*2), one splice site (c.411A>G; p.Gln137Gln), and one non-synonymous (c.1150G>A; p.Gly384Ser) mutation in the ANTXR1 gene. Our studies expand the allelic spectrum in this rare condition and potentially provide insight into the role of ANTXR1 in the regulation of the extracellular matrix.
Keywords: ANTXR1; GAPO syndrome; whole exome sequencing.
© 2014 Wiley Periodicals, Inc.
Conflict of interest statement
Conflict of Interest: J.R.L. has stock ownership in 23 and Me and Ion Torrent Systems, and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from the chromosomal microarray analysis (CMA) and clinical exome sequencing offered in the Medical Genetics Laboratory (MGL;
Figures
References
-
- Aggarwal S, Uttarilli A, Dalal AB. GAPO syndrome with deafness: New feature or incidental finding? Clin Dysmorphol. 2013;22:161–163. - PubMed
-
- Bamshad MJ, Shendure JA, Valle D, Hamosh A, Lupski JR, Gibbs RA, Boerwinkle E, Lifton RP, Gerstein M, Gunel M, Mane S, Nickerson DA Centers for Mendelian G. The Centers for Mendelian Genomics: A new large-scale initiative to identify the genes underlying rare Mendelian conditions. Am J Med Genet Part A. 2012;158A:1523–1525. - PMC - PubMed
-
- Bozkurt B, Yildirim MS, Okka M, Bitirgen G. GAPO syndrome: Four new patients with congenital glaucoma and myelinated retinal nerve fiber layer. Am J Med Genet Part A. 2013;161A:829–834. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
