

Next-generation sequencing identifies rare variants associated with Noonan syndrome
- PMID: 25049390
- PMCID: PMC4128129
- DOI: 10.1073/pnas.1324128111
Next-generation sequencing identifies rare variants associated with Noonan syndrome
Abstract
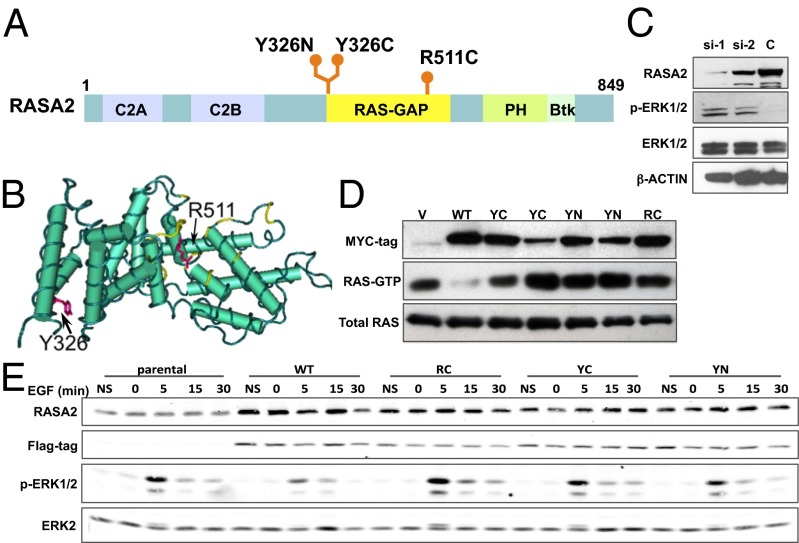
Noonan syndrome (NS) is a relatively common genetic disorder, characterized by typical facies, short stature, developmental delay, and cardiac abnormalities. Known causative genes account for 70-80% of clinically diagnosed NS patients, but the genetic basis for the remaining 20-30% of cases is unknown. We performed next-generation sequencing on germ-line DNA from 27 NS patients lacking a mutation in the known NS genes. We identified gain-of-function alleles in Ras-like without CAAX 1 (RIT1) and mitogen-activated protein kinase kinase 1 (MAP2K1) and previously unseen loss-of-function variants in RAS p21 protein activator 2 (RASA2) that are likely to cause NS in these patients. Expression of the mutant RASA2, MAP2K1, or RIT1 alleles in heterologous cells increased RAS-ERK pathway activation, supporting a causative role in NS pathogenesis. Two patients had more than one disease-associated variant. Moreover, the diagnosis of an individual initially thought to have NS was revised to neurofibromatosis type 1 based on an NF1 nonsense mutation detected in this patient. Another patient harbored a missense mutation in NF1 that resulted in decreased protein stability and impaired ability to suppress RAS-ERK activation; however, this patient continues to exhibit a NS-like phenotype. In addition, a nonsense mutation in RPS6KA3 was found in one patient initially diagnosed with NS whose diagnosis was later revised to Coffin-Lowry syndrome. Finally, we identified other potential candidates for new NS genes, as well as potential carrier alleles for unrelated syndromes. Taken together, our data suggest that next-generation sequencing can provide a useful adjunct to RASopathy diagnosis and emphasize that the standard clinical categories for RASopathies might not be adequate to describe all patients.
Keywords: PTPN11; RAS; developmental diseases; human genetics; whole exome sequencing.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



References
-
- Gelb BD, Tartaglia M. Noonan syndrome and related disorders: Dysregulated RAS-mitogen activated protein kinase signal transduction. Hum Mol Genet. 2006;15(Spec No 2):R220–R226. - PubMed
-
- Aoki Y, Matsubara Y. Ras/MAPK syndromes and childhood hemato-oncological diseases. Int J Hematol. 2013;97(1):30–36. - PubMed
-
- Kontaridis MI, Swanson KD, David FS, Barford D, Neel BG. PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. J Biol Chem. 2006;281(10):6785–6792. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
