

Mutant β-III spectrin causes mGluR1α mislocalization and functional deficits in a mouse model of spinocerebellar ataxia type 5
- PMID: 25057192
- PMCID: PMC4107406
- DOI: 10.1523/JNEUROSCI.0876-14.2014
Mutant β-III spectrin causes mGluR1α mislocalization and functional deficits in a mouse model of spinocerebellar ataxia type 5
Abstract
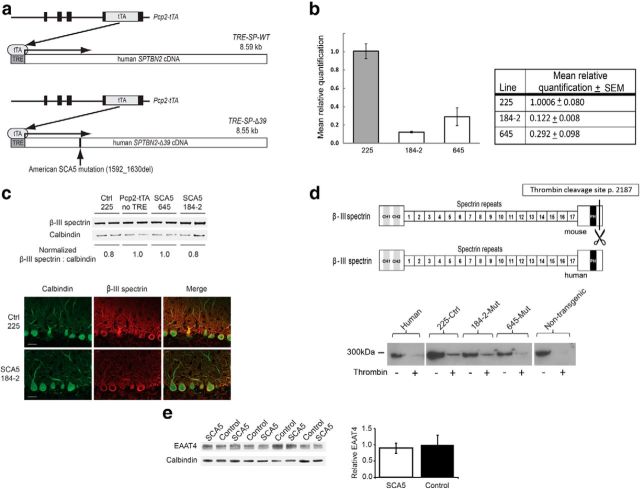
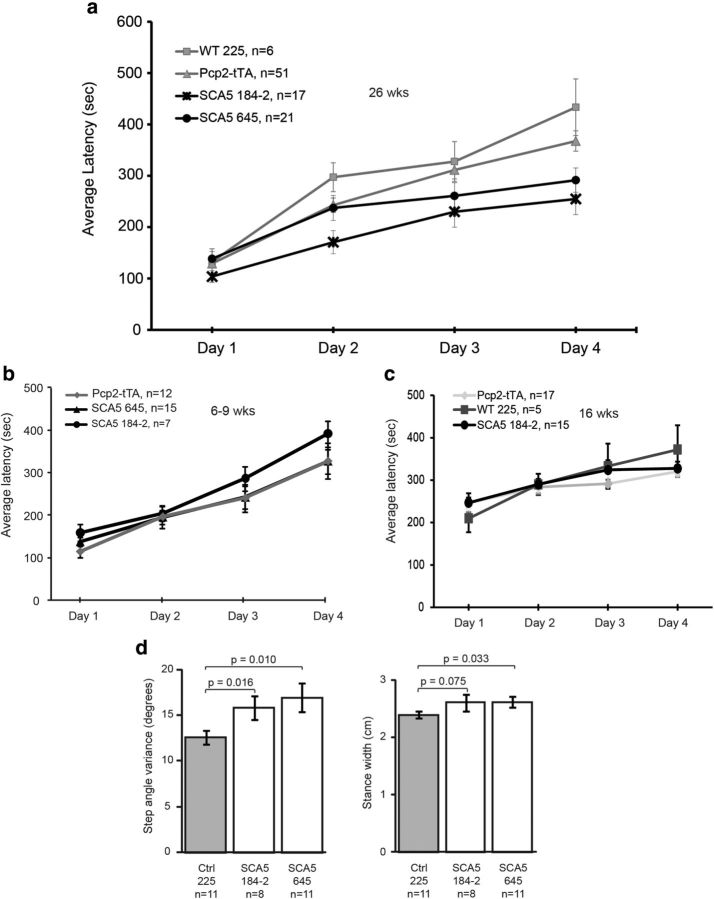
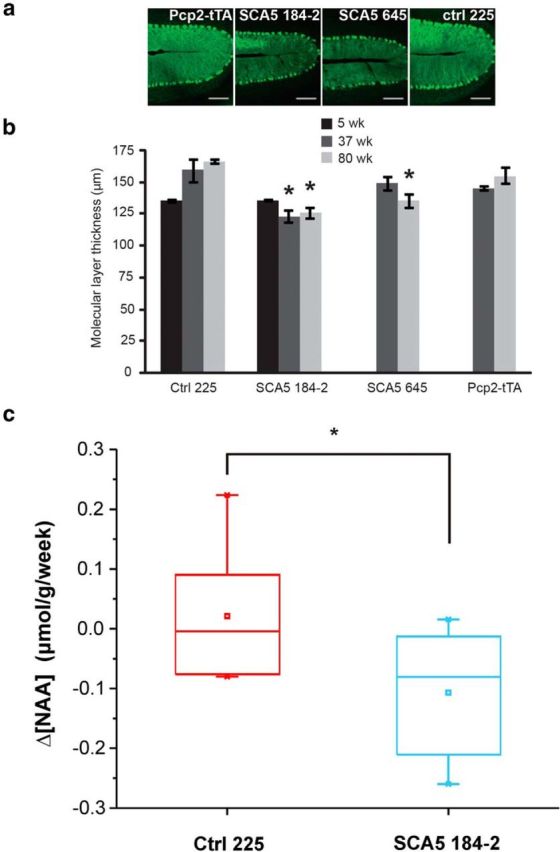
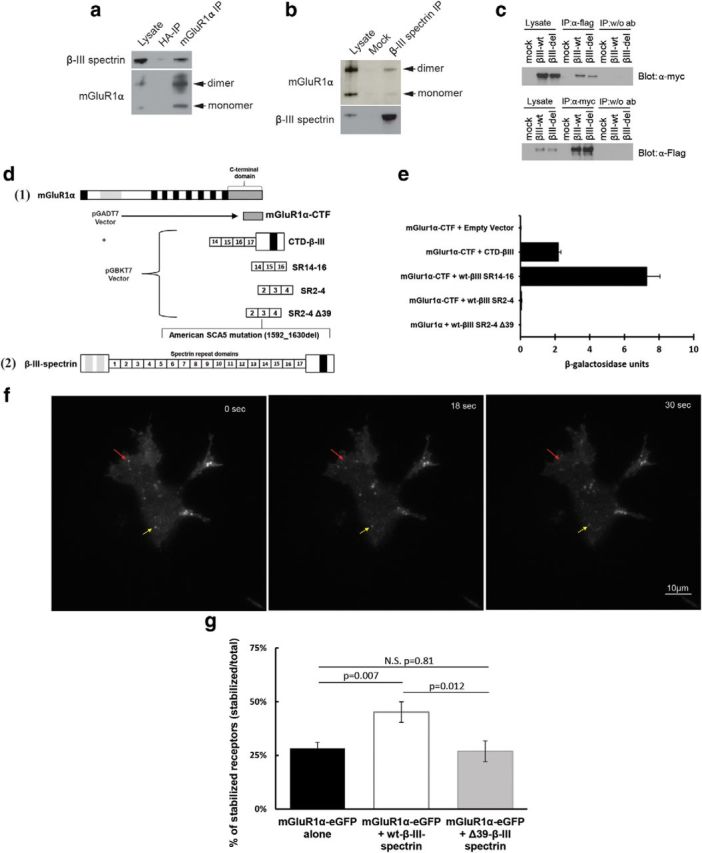
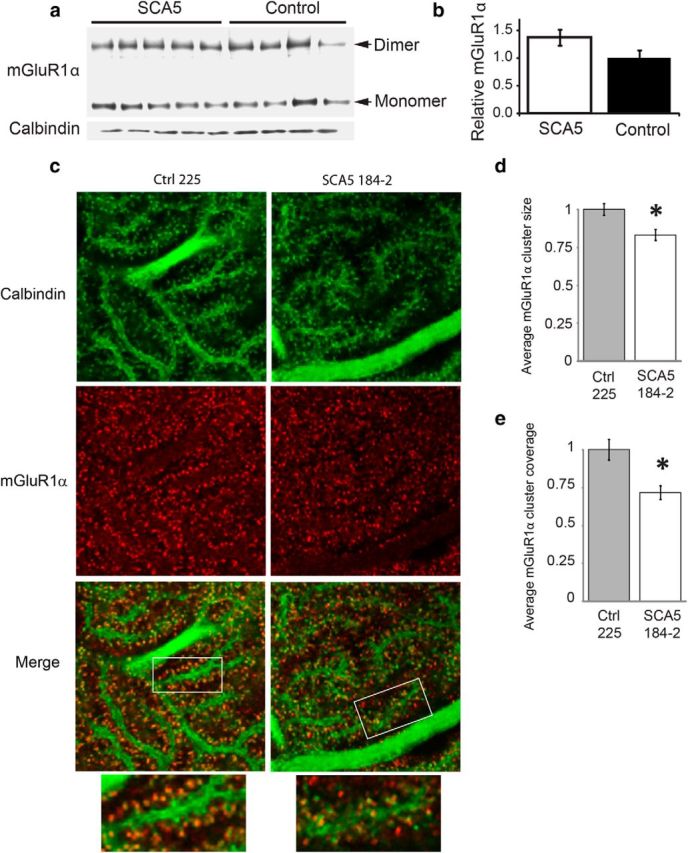
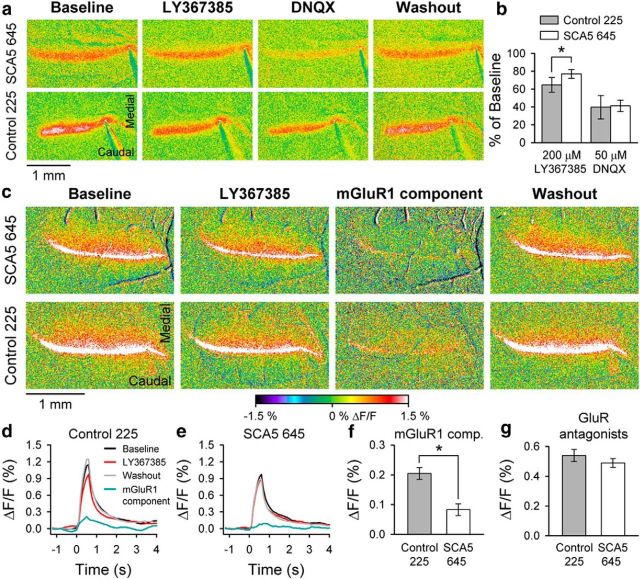
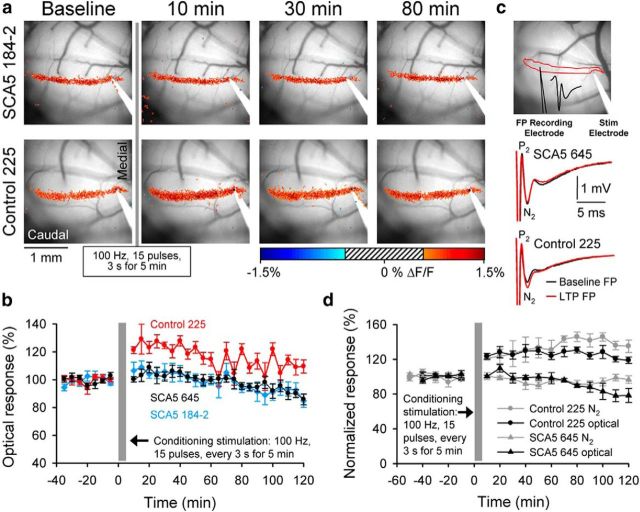
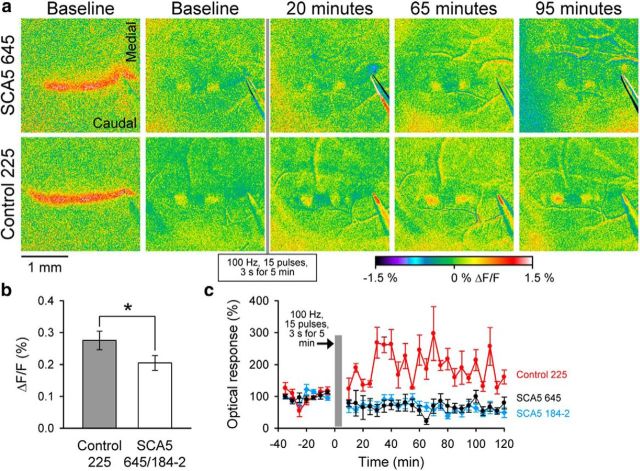
Spinocerebellar ataxia type 5 (SCA5), a dominant neurodegenerative disease characterized by profound Purkinje cell loss, is caused by mutations in SPTBN2, a gene that encodes β-III spectrin. SCA5 is the first neurodegenerative disorder reported to be caused by mutations in a cytoskeletal spectrin gene. We have developed a mouse model to understand the mechanistic basis for this disease and show that expression of mutant but not wild-type β-III spectrin causes progressive motor deficits and cerebellar degeneration. We show that endogenous β-III spectrin interacts with the metabotropic glutamate receptor 1α (mGluR1α) and that mice expressing mutant β-III spectrin have cerebellar dysfunction with altered mGluR1α localization at Purkinje cell dendritic spines, decreased mGluR1-mediated responses, and deficient mGluR1-mediated long-term potentiation. These results indicate that mutant β-III spectrin causes mislocalization and dysfunction of mGluR1α at dendritic spines and connects SCA5 with other disorders involving glutamatergic dysfunction and synaptic plasticity abnormalities.
Keywords: Purkinje cells; long term potentiation; mGluR1α; mouse model; neurodegeneration; spinocerebellar ataxia type 5.
Copyright © 2014 the authors 0270-6474/14/349891-14$15.00/0.
Figures
References
-
- Barnes JA. University of Minnesota; 2011. A functional examination of the afferent cerebellar cortical circuitry of ATXN1 mice in vivo. PhD thesis.
-
- Baude A, Nusser Z, Roberts JD, Mulvihill E, Mcllhinney RA, Somogyi P. The metabotropic glutamate receptor (mGluRl[alpha]) is concentrated at perisynaptic membrane of neuronal subpopulations as detected by immunogold reaction. Neuron. 1993;11:771–787. doi: 10.1016/0896-6273(93)90086-7. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases