

The frustrated host response to Legionella pneumophila is bypassed by MyD88-dependent translation of pro-inflammatory cytokines
- PMID: 25058342
- PMCID: PMC4110041
- DOI: 10.1371/journal.ppat.1004229
The frustrated host response to Legionella pneumophila is bypassed by MyD88-dependent translation of pro-inflammatory cytokines
Abstract
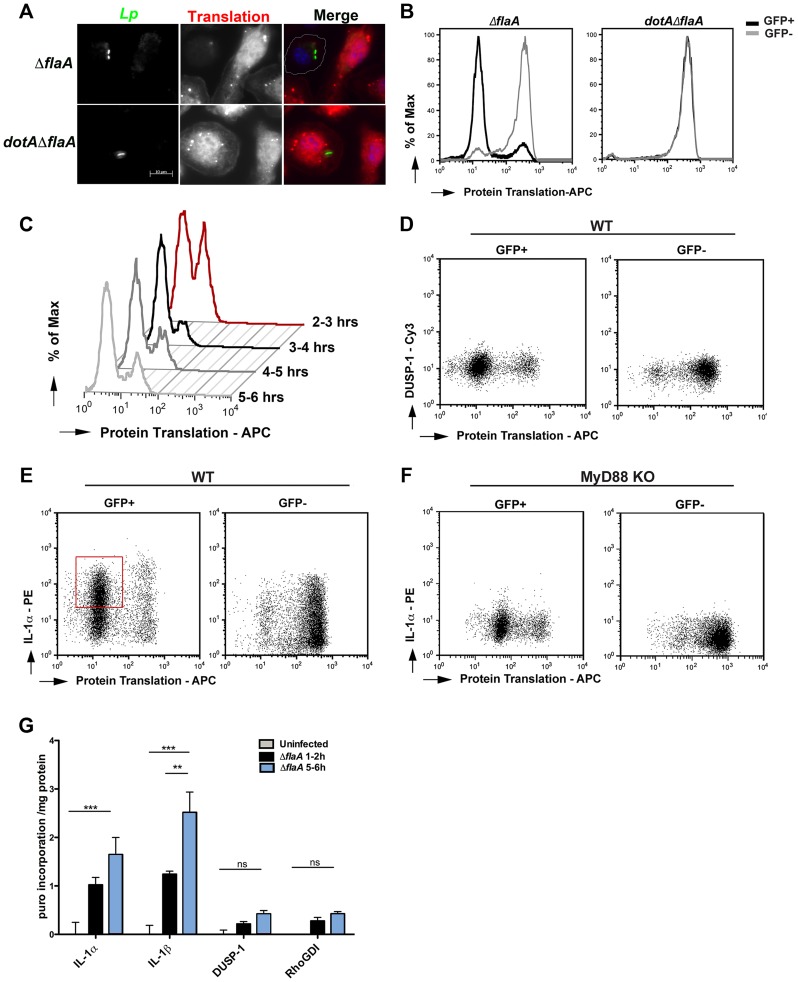
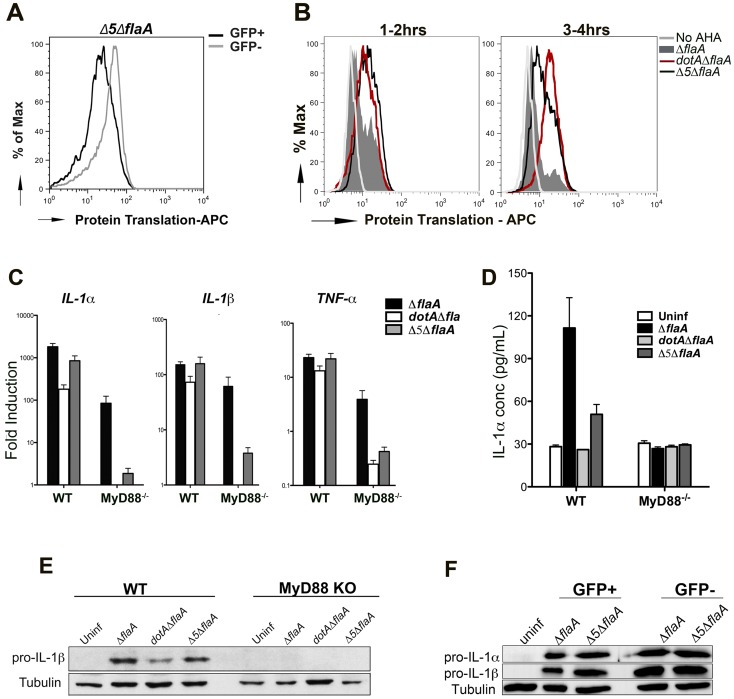
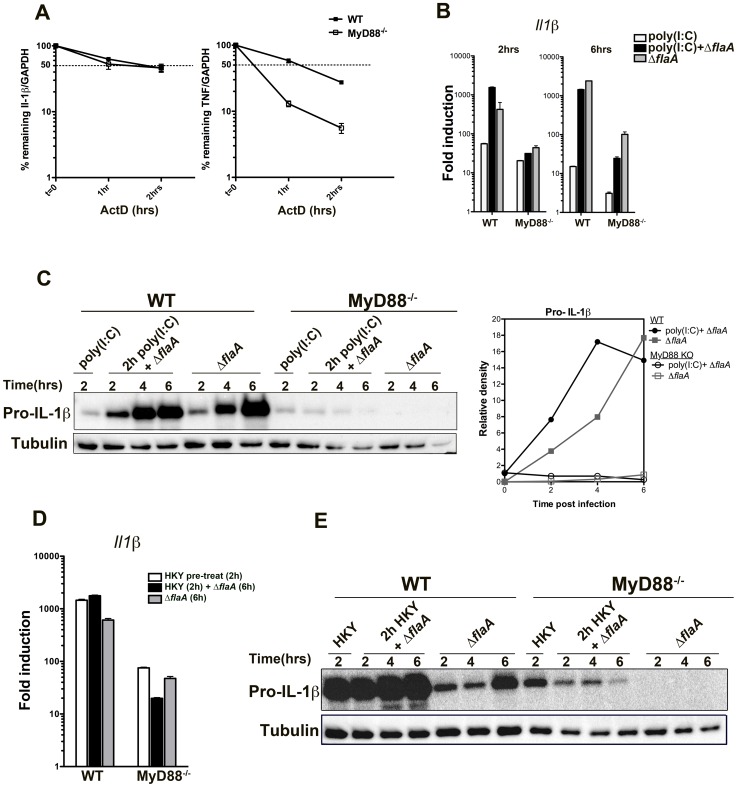
Many pathogens, particularly those that require their host for survival, have devised mechanisms to subvert the host immune response in order to survive and replicate intracellularly. Legionella pneumophila, the causative agent of Legionnaires' disease, promotes intracellular growth by translocating proteins into its host cytosol through its type IV protein secretion machinery. At least 5 of the bacterial translocated effectors interfere with the function of host cell elongation factors, blocking translation and causing the induction of a unique host cell transcriptional profile. In addition, L. pneumophila also interferes with translation initiation, by preventing cap-dependent translation in host cells. We demonstrate here that protein translation inhibition by L. pneumophila leads to a frustrated host MAP kinase response, where genes involved in the pathway are transcribed but fail to be translated due to the bacterium-induced protein synthesis inhibition. Surprisingly, few pro-inflammatory cytokines, such as IL-1α and IL-1β, bypass this inhibition and get synthesized in the presence of Legionella effectors. We show that the selective synthesis of these genes requires MyD88 signaling and takes place in both infected cells that harbor bacteria and neighboring bystander cells. Our findings offer a perspective of how host cells are able to cope with pathogen-encoded activities that disrupt normal cellular process and initiate a successful inflammatory response.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
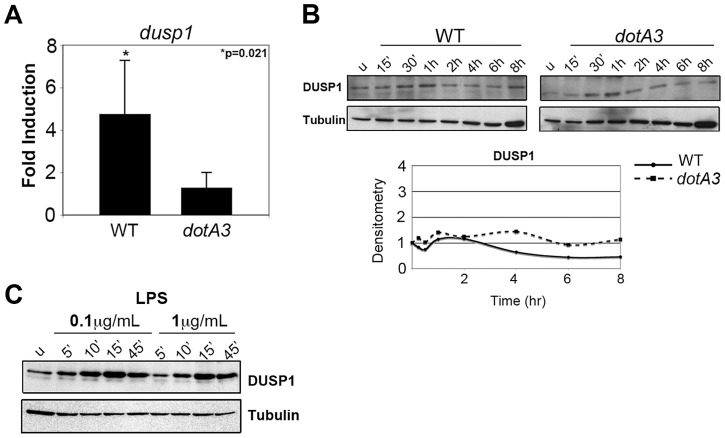
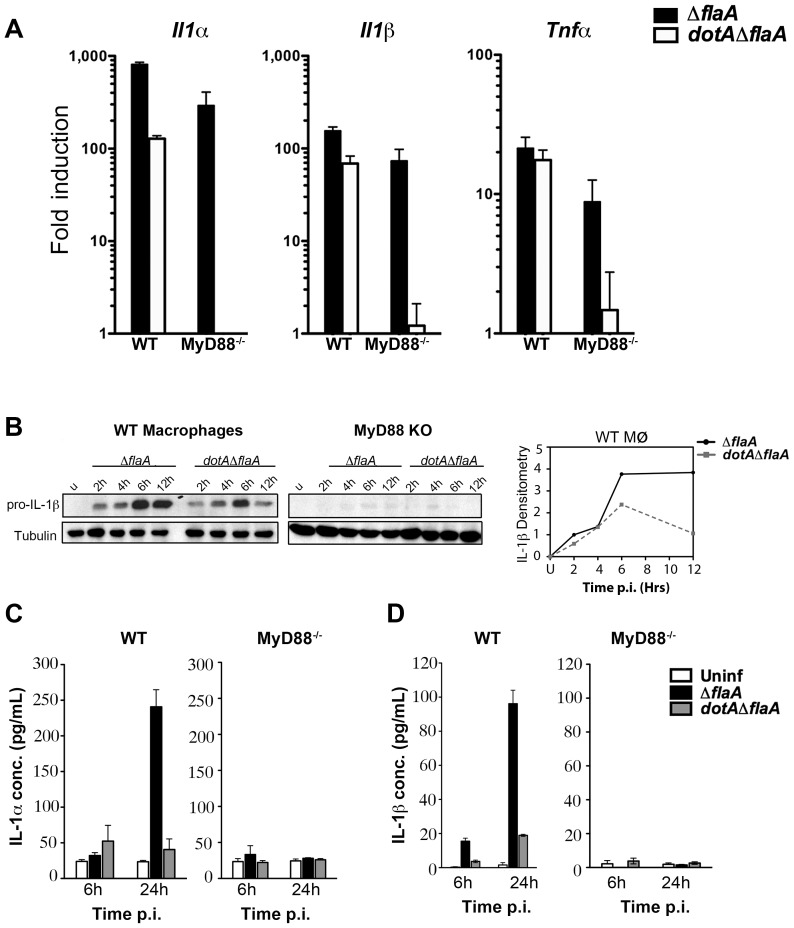
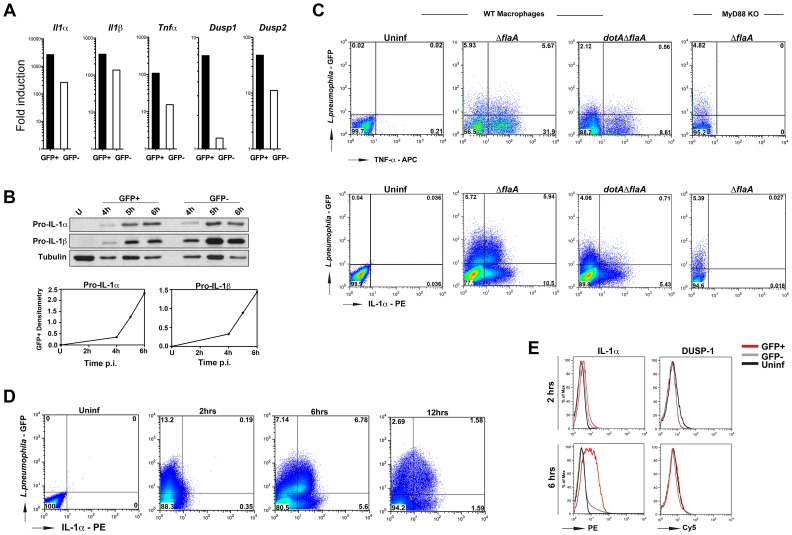
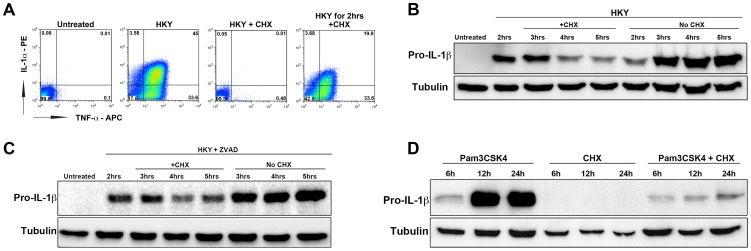
References
-
- Akira S, Hemmi H (2003) Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett 85: 85–95. - PubMed
-
- Medzhitov R, Janeway C Jr (2000) Innate immunity. N Engl J Med 343: 338–344. - PubMed
-
- Jones JD, Dangl JL (2006) The plant immune system. Nature 444: 323–329. - PubMed
-
- Dangl JL, Jones JD (2001) Plant pathogens and integrated defence responses to infection. Nature 411: 826–833. - PubMed
-
- Ausubel FM (2005) Are innate immune signaling pathways in plants and animals conserved? Nat Immunol 6: 973–979. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
