

Multiscale modeling of biological functions: from enzymes to molecular machines (Nobel Lecture)
- PMID: 25060243
- PMCID: PMC4948593
- DOI: 10.1002/anie.201403689
Multiscale modeling of biological functions: from enzymes to molecular machines (Nobel Lecture)
Abstract
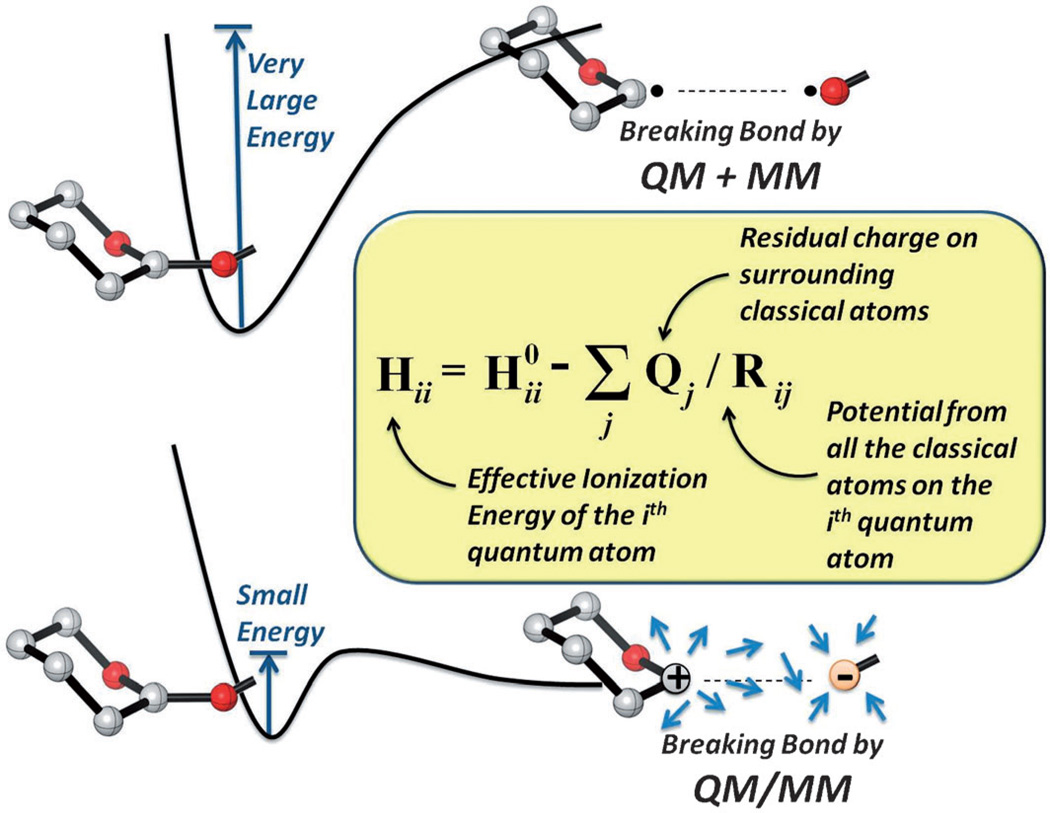
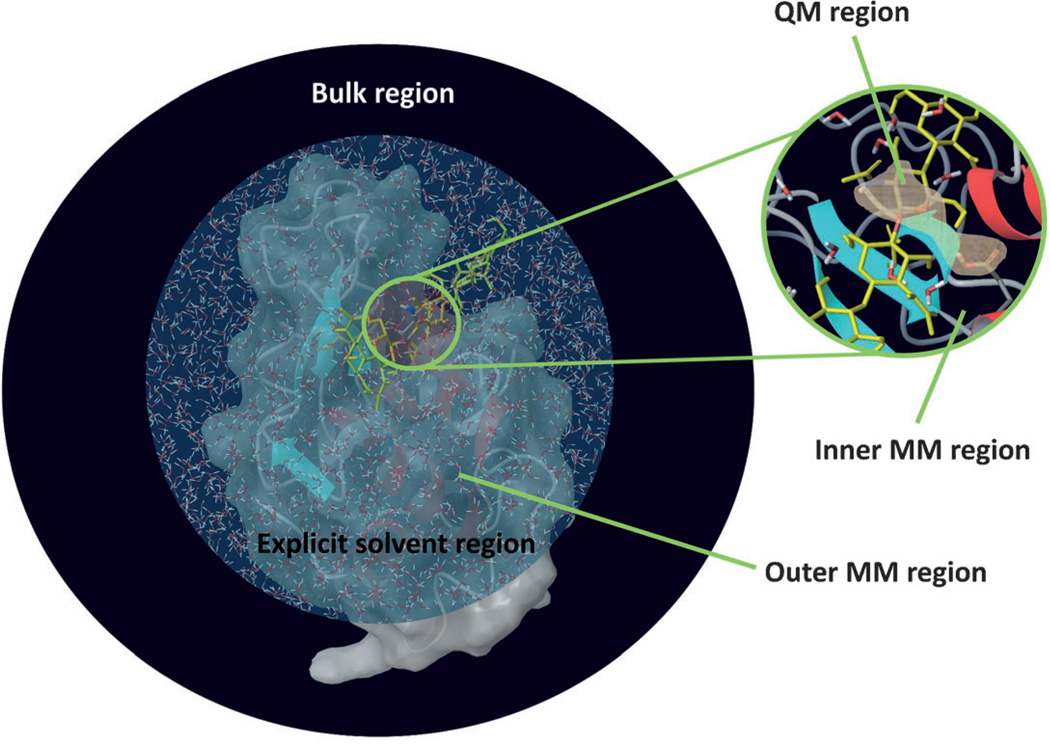
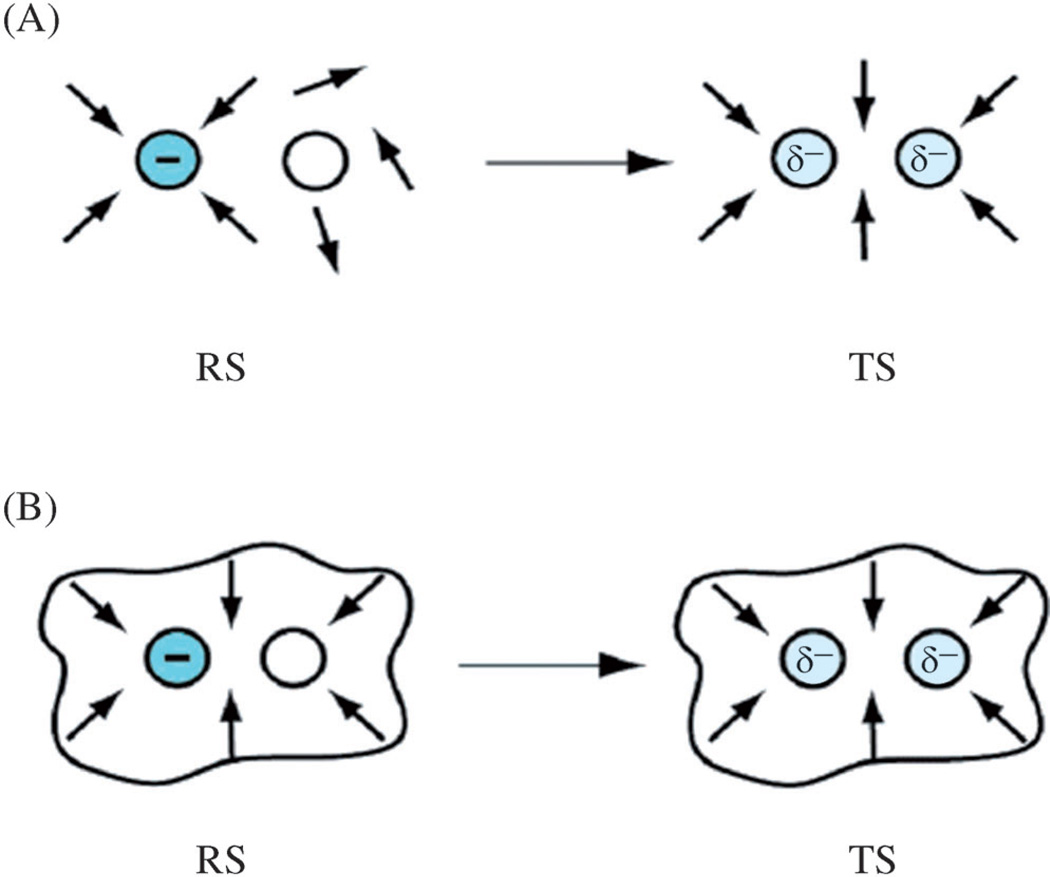
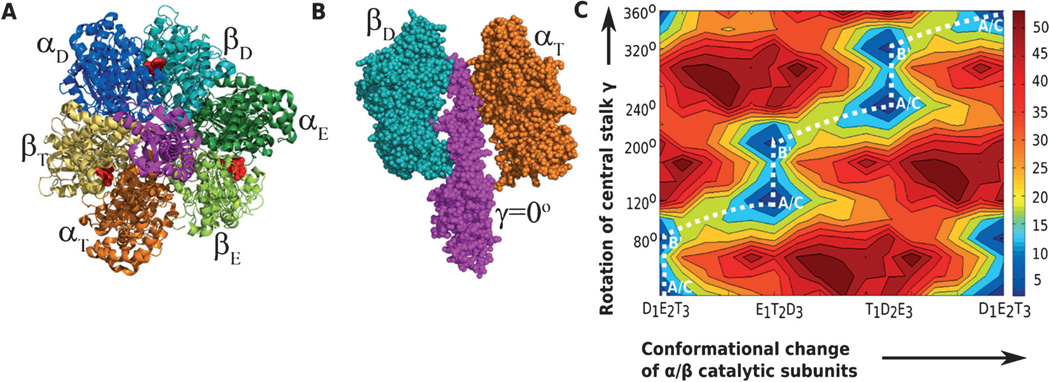
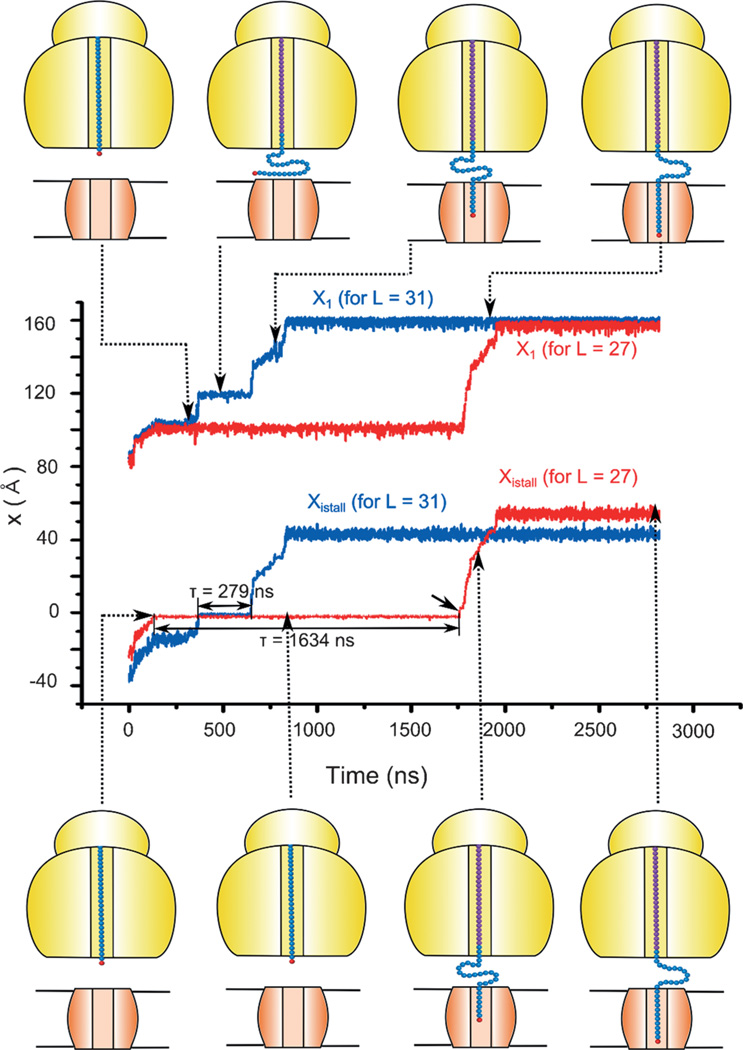
A detailed understanding of the action of biological molecules is a pre-requisite for rational advances in health sciences and related fields. Here, the challenge is to move from available structural information to a clear understanding of the underlying function of the system. In light of the complexity of macromolecular complexes, it is essential to use computer simulations to describe how the molecular forces are related to a given function. However, using a full and reliable quantum mechanical representation of large molecular systems has been practically impossible. The solution to this (and related) problems has emerged from the realization that large systems can be spatially divided into a region where the quantum mechanical description is essential (e.g. a region where bonds are being broken), with the remainder of the system being represented on a simpler level by empirical force fields. This idea has been particularly effective in the development of the combined quantum mechanics/molecular mechanics (QM/MM) models. Here, the coupling between the electrostatic effects of the quantum and classical subsystems has been a key to the advances in describing the functions of enzymes and other biological molecules. The same idea of representing complex systems in different resolutions in both time and length scales has been found to be very useful in modeling the action of complex systems. In such cases, starting with coarse grained (CG) representations that were originally found to be very useful in simulating protein folding, and augmenting them with a focus on electrostatic energies, has led to models that are particularly effective in probing the action of molecular machines. The same multiscale idea is likely to play a major role in modeling of even more complex systems, including cells and collections of cells.
Keywords: biomolecules; computational chemistry; free energy calculations; molecular modeling; multiscale models.
© 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Figures
References
-
-
Quantum Chemical Models (Nobel Lecture): Pople JA. Angew. Chem. 1999;111:2014–2023. Angew. Chem. Int. Ed. 1999;38:1894–1902.
-
-
-
Theoretical Studies of Enzymic Reactions – Dielectric, Electrostatic and Steric Stabilization of Carbonium-Ion in Reaction of Lysozyme: Warshel A, Levitt M. J. Mol. Biol. 1976;103:227–249.
-
-
- Fersht A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. New York: Freeman; 1999. p. 631.
-
- Warshel A. Computer Modeling of Chemical Reactions in Enzymes and Solutions. Hoboken: Wiley-Interscience; 1997. p. 256.
-
- Jencks WP. Catalysis in Chemistry and Enzymology. New York: McGraw-Hill; 1969. p. 644.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
