

Appearances can be deceptive: revealing a hidden viral infection with deep sequencing in a plant quarantine context
- PMID: 25061967
- PMCID: PMC4111361
- DOI: 10.1371/journal.pone.0102945
Appearances can be deceptive: revealing a hidden viral infection with deep sequencing in a plant quarantine context
Abstract
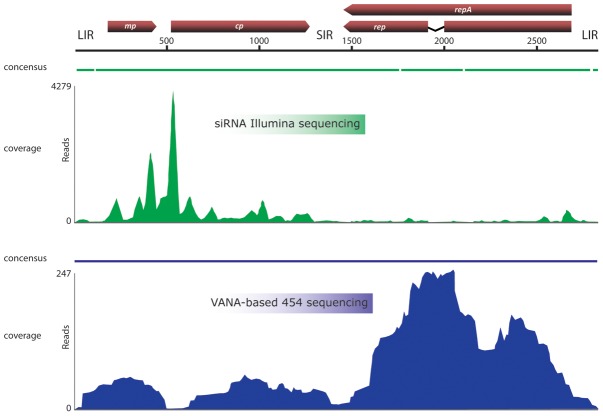
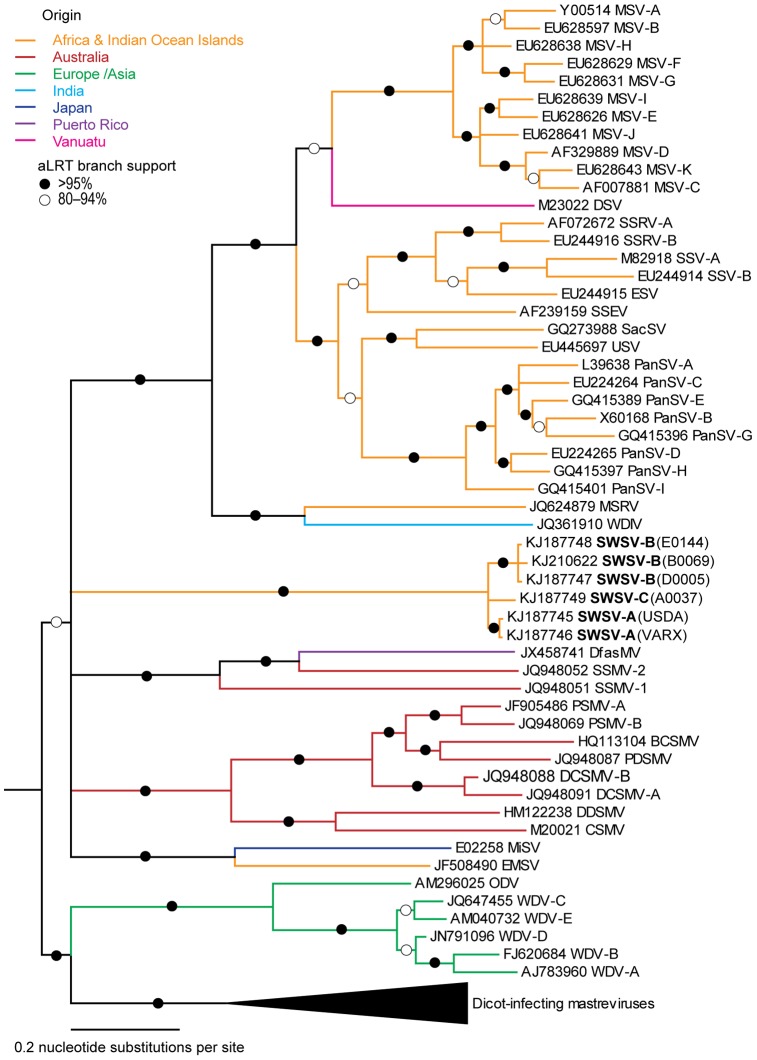
Comprehensive inventories of plant viral diversity are essential for effective quarantine and sanitation efforts. The safety of regulated plant material exchanges presently relies heavily on techniques such as PCR or nucleic acid hybridisation, which are only suited to the detection and characterisation of specific, well characterised pathogens. Here, we demonstrate the utility of sequence-independent next generation sequencing (NGS) of both virus-derived small interfering RNAs (siRNAs) and virion-associated nucleic acids (VANA) for the detailed identification and characterisation of viruses infecting two quarantined sugarcane plants. Both plants originated from Egypt and were known to be infected with Sugarcane streak Egypt Virus (SSEV; Genus Mastrevirus, Family Geminiviridae), but were revealed by the NGS approaches to also be infected by a second highly divergent mastrevirus, here named Sugarcane white streak Virus (SWSV). This novel virus had escaped detection by all routine quarantine detection assays and was found to also be present in sugarcane plants originating from Sudan. Complete SWSV genomes were cloned and sequenced from six plants and all were found to share >91% genome-wide identity. With the exception of two SWSV variants, which potentially express unusually large RepA proteins, the SWSV isolates display genome characteristics very typical to those of all other previously described mastreviruses. An analysis of virus-derived siRNAs for SWSV and SSEV showed them to be strongly influenced by secondary structures within both genomic single stranded DNA and mRNA transcripts. In addition, the distribution of siRNA size frequencies indicates that these mastreviruses are likely subject to both transcriptional and post-transcriptional gene silencing. Our study stresses the potential advantages of NGS-based virus metagenomic screening in a plant quarantine setting and indicates that such techniques could dramatically reduce the numbers of non-intercepted virus pathogens passing through plant quarantine stations.
Conflict of interest statement
Figures


References
-
- Anderson PK, Cunningham AA, Patel NG, Morales FJ, Epstein PR, et al. (2004) Emerging infectious diseases of plants: pathogen pollution, climate change and agrotechnology drivers. Trends Ecol Evol 19: 535–544. - PubMed
-
- Jones RAC (2009) Plant virus emergence and evolution: Origins, new encounter scenarios, factors driving emergence, effects of changing world conditions, and prospects for control. Virus Research 141: 113–130. - PubMed
-
- Roossinck MJ (2011) The good viruses: viral mutualistic symbioses. Nat Rev Microbiol 9: 99–108. - PubMed
-
- Rosario K, Breitbart M (2011) Exploring the viral world through metagenomics. Curr Opin Virol 1: 1–9. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
