

Purifying selection in deeply conserved human enhancers is more consistent than in coding sequences
- PMID: 25062004
- PMCID: PMC4111549
- DOI: 10.1371/journal.pone.0103357
Purifying selection in deeply conserved human enhancers is more consistent than in coding sequences
Abstract
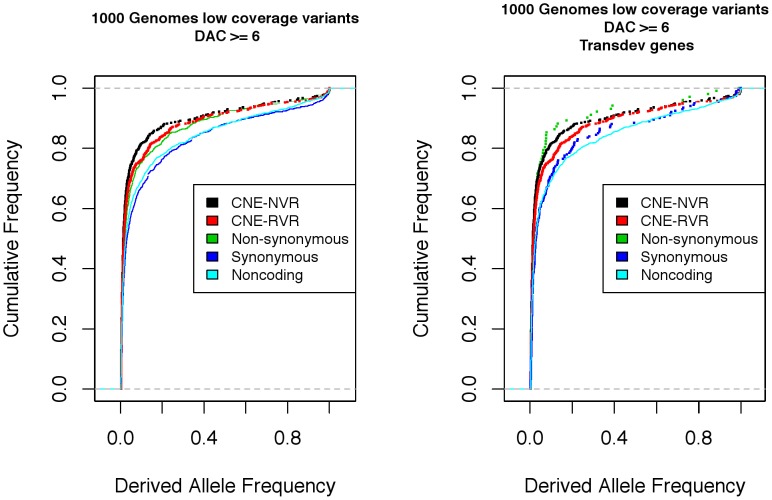
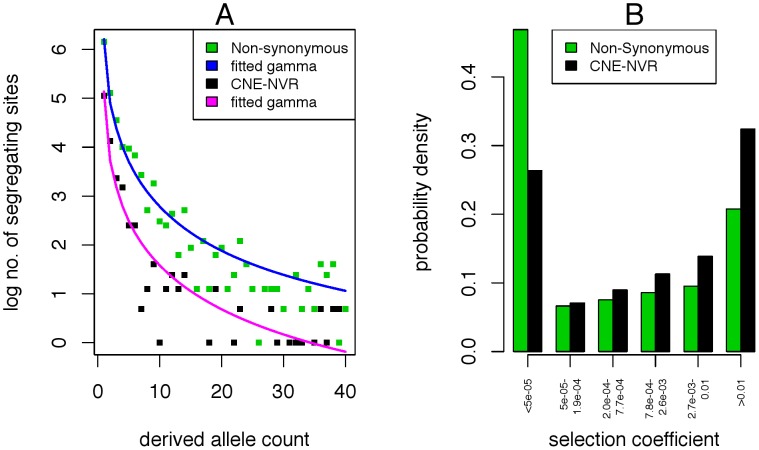
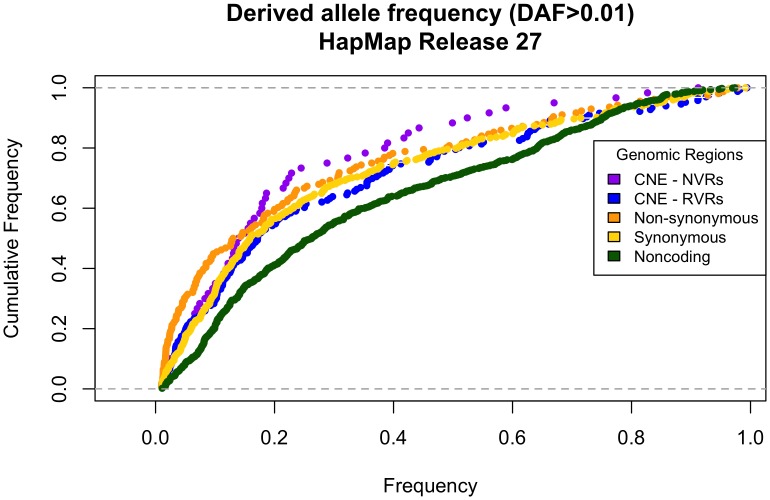
Comparison of polymorphism at synonymous and non-synonymous sites in protein-coding DNA can provide evidence for selective constraint. Non-coding DNA that forms part of the regulatory landscape presents more of a challenge since there is not such a clear-cut distinction between sites under stronger and weaker selective constraint. Here, we consider putative regulatory elements termed Conserved Non-coding Elements (CNEs) defined by their high level of sequence identity across all vertebrates. Some mutations in these regions have been implicated in developmental disorders; we analyse CNE polymorphism data to investigate whether such deleterious effects are widespread in humans. Single nucleotide variants from the HapMap and 1000 Genomes Projects were mapped across nearly 2000 CNEs. In the 1000 Genomes data we find a significant excess of rare derived alleles in CNEs relative to coding sequences; this pattern is absent in HapMap data, apparently obscured by ascertainment bias. The distribution of polymorphism within CNEs is not uniform; we could identify two categories of sites by exploiting deep vertebrate alignments: stretches that are non-variant, and those that have at least one substitution. The conserved category has fewer polymorphic sites and a greater excess of rare derived alleles, which can be explained by a large proportion of sites under strong purifying selection within humans--higher than that for non-synonymous sites in most protein coding regions, and comparable to that at the strongly conserved trans-dev genes. Conversely, the more evolutionarily labile CNE sites have an allele frequency distribution not significantly different from non-synonymous sites. Future studies should exploit genome-wide re-sequencing to obtain better coverage in selected non-coding regions, given the likelihood that mutations in evolutionarily conserved enhancer sequences are deleterious. Discovery pipelines should validate non-coding variants to aid in identifying causal and risk-enhancing variants in complex disorders, in contrast to the current focus on exome sequencing.
Conflict of interest statement
Figures
References
-
- Lettice LA, Heaney SJ, Purdie LA, Li L, de Beer P, et al. (2003) A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet 12: 1725–1735. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
