

Local order in the unfolded state: conformational biases and nearest neighbor interactions
- PMID: 25062017
- PMCID: PMC4192670
- DOI: 10.3390/biom4030725
Local order in the unfolded state: conformational biases and nearest neighbor interactions
Abstract
The discovery of Intrinsically Disordered Proteins, which contain significant levels of disorder yet perform complex biologically functions, as well as unwanted aggregation, has motivated numerous experimental and theoretical studies aimed at describing residue-level conformational ensembles. Multiple lines of evidence gathered over the last 15 years strongly suggest that amino acids residues display unique and restricted conformational preferences in the unfolded state of peptides and proteins, contrary to one of the basic assumptions of the canonical random coil model. To fully understand residue level order/disorder, however, one has to gain a quantitative, experimentally based picture of conformational distributions and to determine the physical basis underlying residue-level conformational biases. Here, we review the experimental, computational and bioinformatic evidence for conformational preferences of amino acid residues in (mostly short) peptides that can be utilized as suitable model systems for unfolded states of peptides and proteins. In this context particular attention is paid to the alleged high polyproline II preference of alanine. We discuss how these conformational propensities may be modulated by peptide solvent interactions and so called nearest-neighbor interactions. The relevance of conformational propensities for the protein folding problem and the understanding of IDPs is briefly discussed.
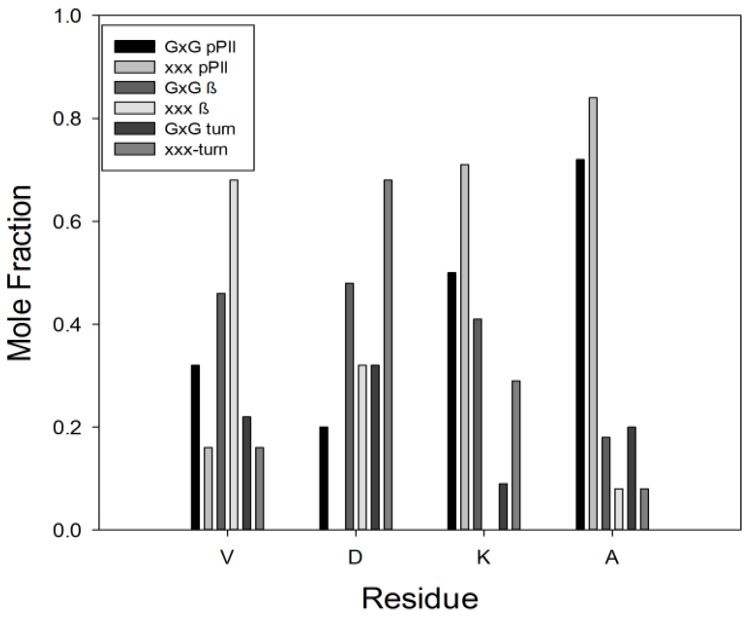
Figures
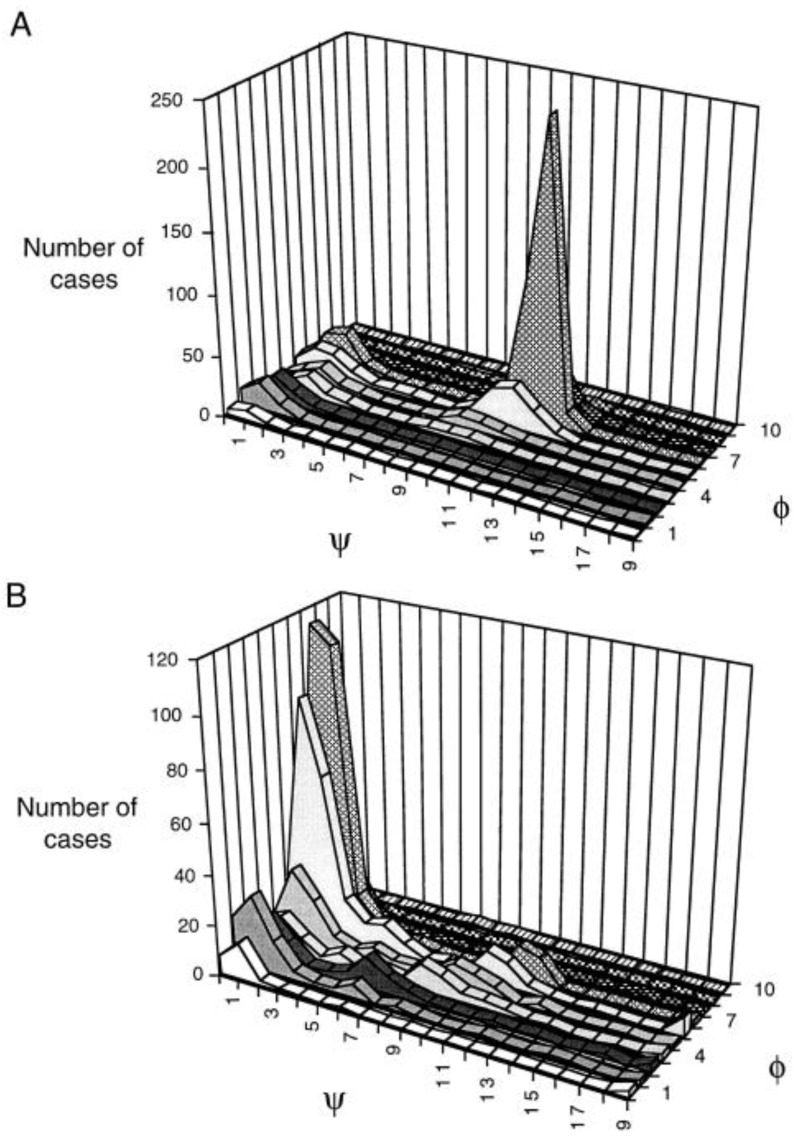
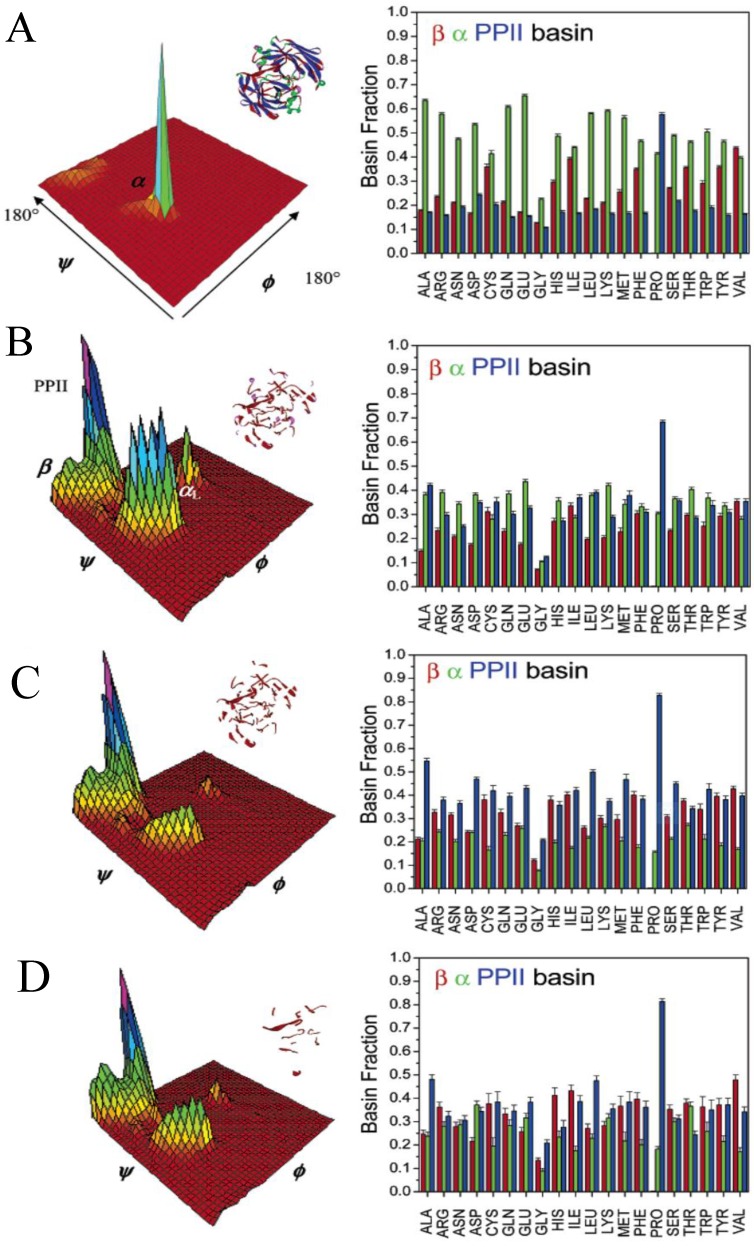
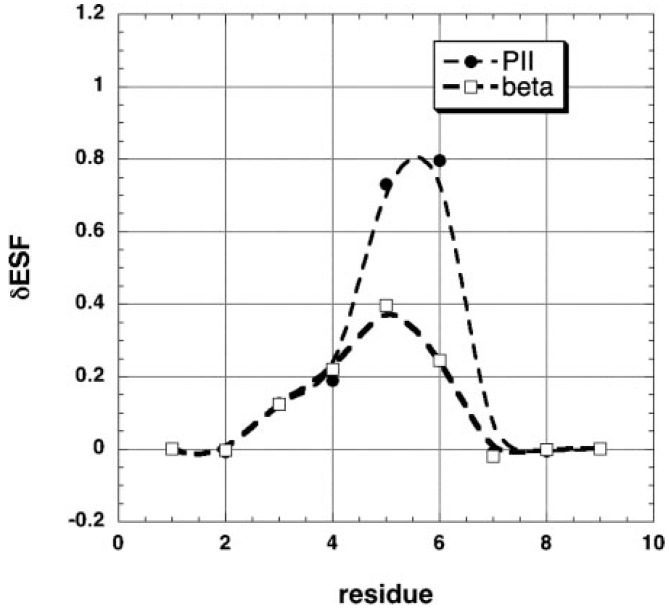
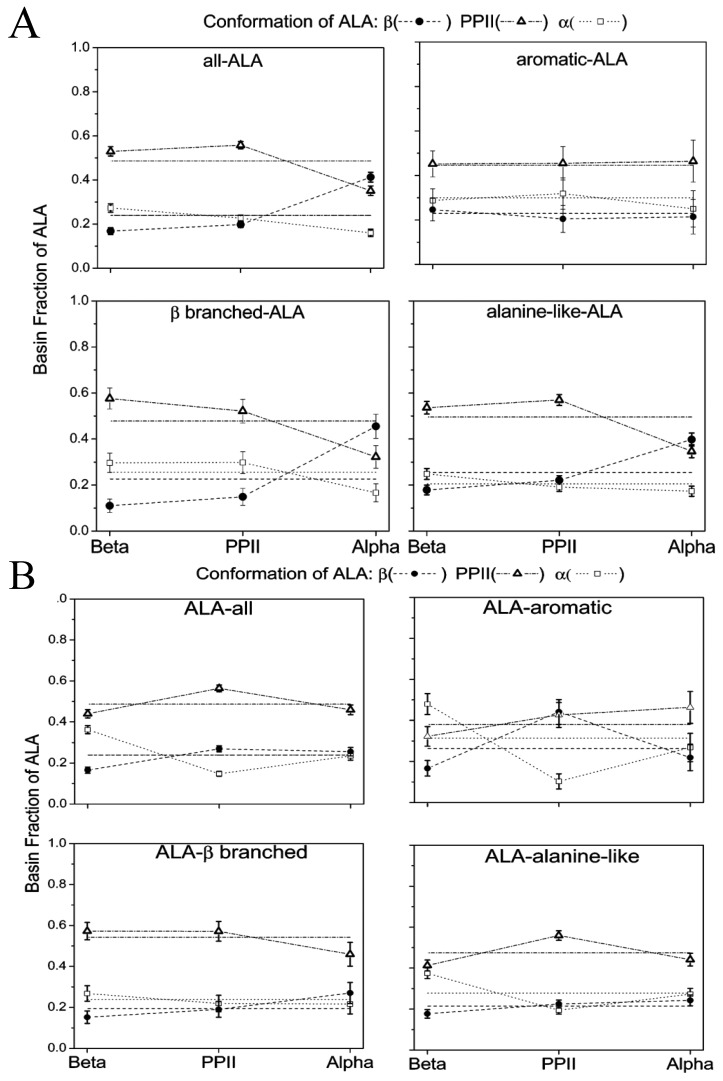
Similar articles
-
Construction and comparison of the statistical coil states of unfolded and intrinsically disordered proteins from nearest-neighbor corrected conformational propensities of short peptides.Mol Biosyst. 2016 Oct 18;12(11):3294-3306. doi: 10.1039/c6mb00489j. Mol Biosyst. 2016. PMID: 27545097
-
Conformational propensities and residual structures in unfolded peptides and proteins.Mol Biosyst. 2012 Jan;8(1):122-33. doi: 10.1039/c1mb05225j. Epub 2011 Aug 30. Mol Biosyst. 2012. PMID: 21879108 Review.
-
Randomizing the unfolded state of peptides (and proteins) by nearest neighbor interactions between unlike residues.Chemistry. 2015 Mar 23;21(13):5173-92. doi: 10.1002/chem.201406539. Epub 2015 Feb 26. Chemistry. 2015. PMID: 25728043
-
Randomizing of Oligopeptide Conformations by Nearest Neighbor Interactions between Amino Acid Residues.Biomolecules. 2022 May 11;12(5):684. doi: 10.3390/biom12050684. Biomolecules. 2022. PMID: 35625612 Free PMC article.
-
Exploring Nearest Neighbor Interactions and Their Influence on the Gibbs Energy Landscape of Unfolded Proteins and Peptides.Int J Mol Sci. 2022 May 18;23(10):5643. doi: 10.3390/ijms23105643. Int J Mol Sci. 2022. PMID: 35628453 Free PMC article. Review.
Cited by
-
Elastin-like polypeptides as models of intrinsically disordered proteins.FEBS Lett. 2015 Sep 14;589(19 Pt A):2477-86. doi: 10.1016/j.febslet.2015.08.029. Epub 2015 Aug 29. FEBS Lett. 2015. PMID: 26325592 Free PMC article. Review.
-
Unfavorable regions in the ramachandran plot: Is it really steric hindrance? The interacting quantum atoms perspective.J Comput Chem. 2017 Nov 5;38(29):2459-2474. doi: 10.1002/jcc.24904. Epub 2017 Aug 25. J Comput Chem. 2017. PMID: 28841241 Free PMC article.
-
Reduced alphabet of prebiotic amino acids optimally encodes the conformational space of diverse extant protein folds.BMC Evol Biol. 2019 Jul 30;19(1):158. doi: 10.1186/s12862-019-1464-6. BMC Evol Biol. 2019. PMID: 31362700 Free PMC article.
-
Using NMR Chemical Shifts to Determine Residue-Specific Secondary Structure Populations for Intrinsically Disordered Proteins.Methods Enzymol. 2018;611:101-136. doi: 10.1016/bs.mie.2018.09.011. Epub 2018 Oct 22. Methods Enzymol. 2018. PMID: 30471686 Free PMC article.
-
Revisiting the Formation of a Native Disulfide Bond: Consequences for Protein Regeneration and Beyond.Molecules. 2020 Nov 16;25(22):5337. doi: 10.3390/molecules25225337. Molecules. 2020. PMID: 33207635 Free PMC article. Review.
References
-
- Uversky V., Dunker A.K. Why are we interested in the unfolded peptides and proteins? In: Schweitzer-Stenner R., editor. Protein and Peptide Folding, Misfolding, and Non-folding. Wiley & Sons; Hoboken, NJ, USA: 2012. pp. 3–54.
-
- Ben Naim A. The role of hydrogen bonds in protein folding and protein association. J. Phys. Chem. 1991;95:1437–1444. doi: 10.1021/j100156a074. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
