

Rapid whole-genome sequencing for surveillance of Salmonella enterica serovar enteritidis
- PMID: 25062035
- PMCID: PMC4111163
- DOI: 10.3201/eid2008.131399
Rapid whole-genome sequencing for surveillance of Salmonella enterica serovar enteritidis
Abstract
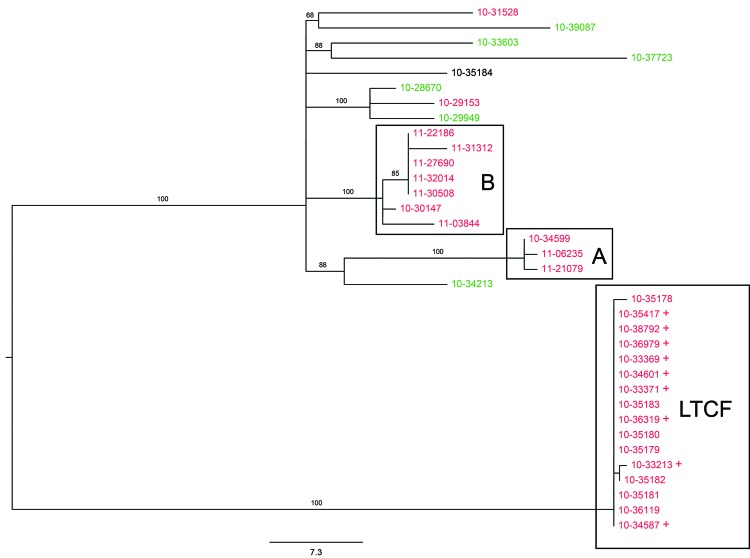
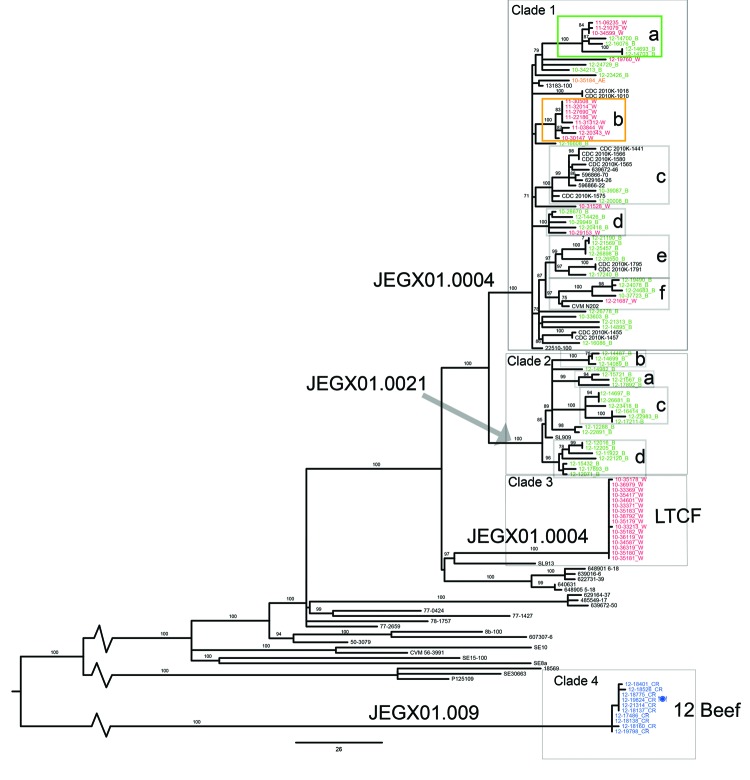
For Salmonella enterica serovar Enteritidis, 85% of isolates can be classified into 5 pulsed-field gel electrophoresis (PFGE) types. However, PFGE has limited discriminatory power for outbreak detection. Although whole-genome sequencing has been found to improve discrimination of outbreak clusters, whether this procedure can be used in real-time in a public health laboratory is not known. Therefore, we conducted a retrospective and prospective analysis. The retrospective study investigated isolates from 1 confirmed outbreak. Additional cases could be attributed to the outbreak strain on the basis of whole-genome data. The prospective study included 58 isolates obtained in 2012, including isolates from 1 epidemiologically defined outbreak. Whole-genome sequencing identified additional isolates that could be attributed to the outbreak, but which differed from the outbreak-associated PFGE type. Additional putative outbreak clusters were detected in the retrospective and prospective analyses. This study demonstrates the practicality of implementing this approach for outbreak surveillance in a state public health laboratory.
Keywords: Salmonella enterica serovar Enteritidis; bacteria; high-throughput nucleotide sequencing; infectious disease outbreaks; public health laboratory surveillance; pulsed-field gel electrophoresis; whole-genome sequencing.
Figures
References
-
- Bakker HC, Switt AI, Cummings CA, Hoelzer K, Degoricija L, Rodriguez-Rivera LD, et al. A whole genome SNP based approach to trace and identify outbreaks linked to a common Salmonella enterica subsp. enterica serovar Montevideo pulsed-field gel electrophoresis type. Appl Environ Microbiol. 2011;77:8648–55. 10.1128/AEM.06538-11 - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
