

Retinoblastoma. Fifty years of progress. The LXXI Edward Jackson Memorial Lecture
- PMID: 25065496
- PMCID: PMC4250440
- DOI: 10.1016/j.ajo.2014.07.025
Retinoblastoma. Fifty years of progress. The LXXI Edward Jackson Memorial Lecture
Abstract
Purpose: To review the progress made in understanding the genetic basis, molecular pathology, and treatment of retinoblastoma since the previous Jackson lecture on the topic was published 50 years ago.
Design: Perspective based on personal experience and the literature.
Methods: The literature regarding retinoblastoma was reviewed since 1963. Advances in understanding the biology and treatment of retinoblastoma provided context through the author's clinical, pathologic, and research experiences.
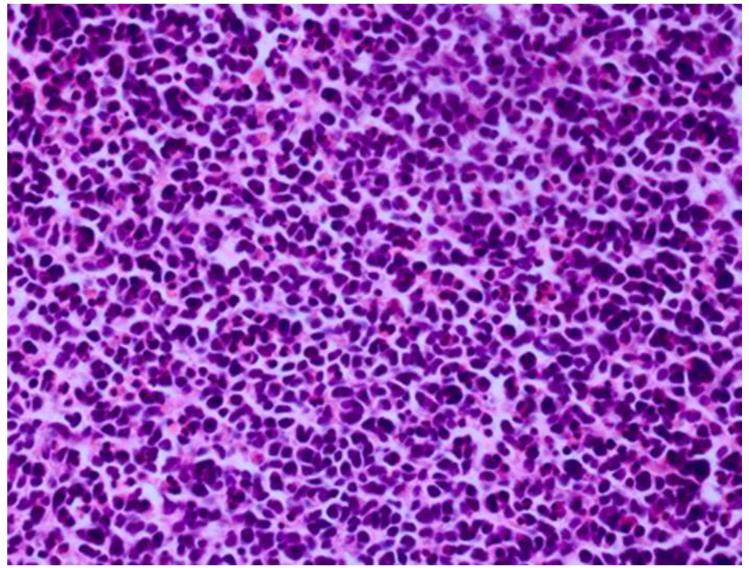
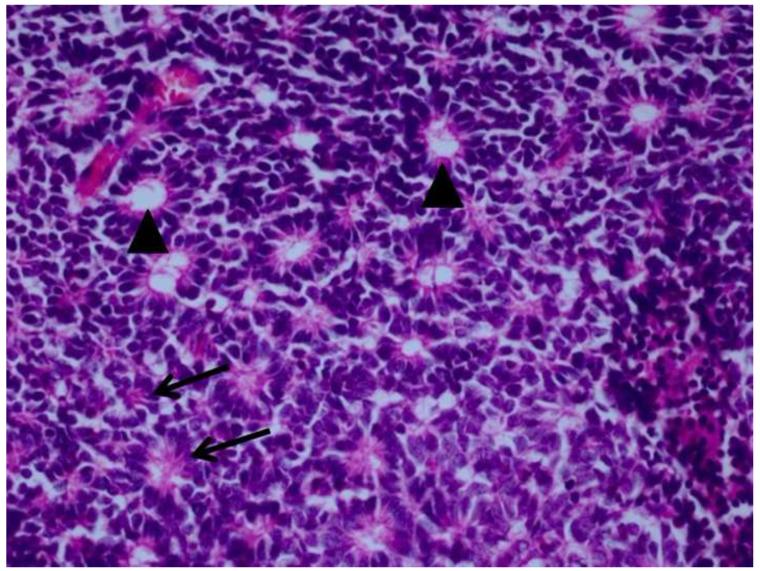
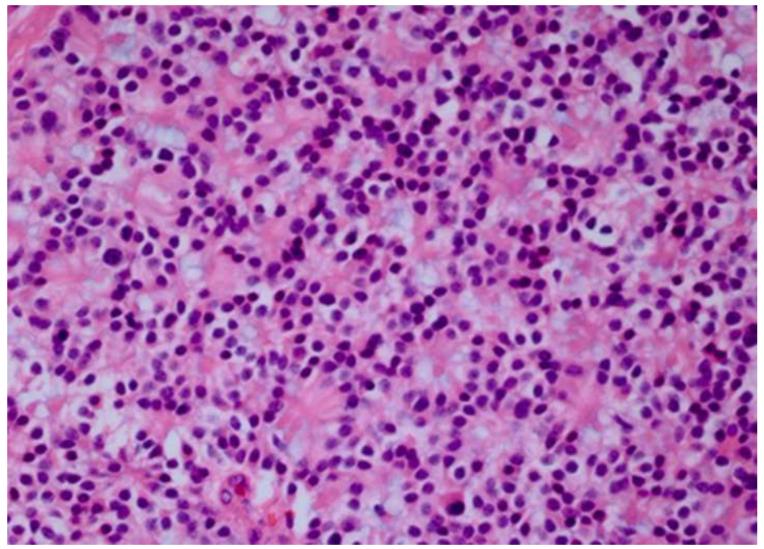
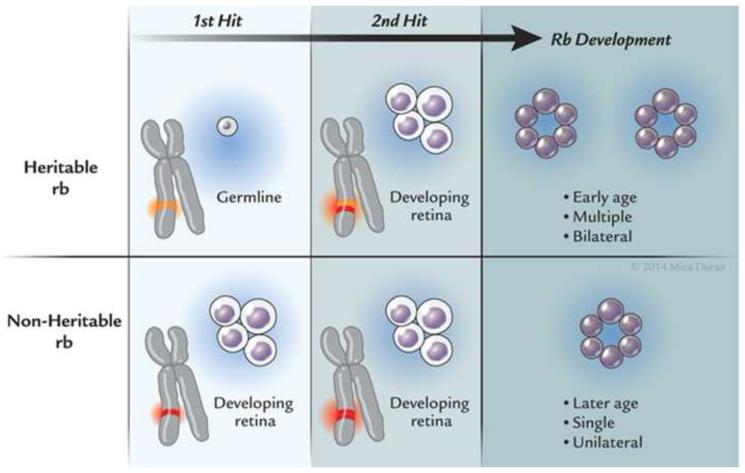
Results: Retinoblastoma was first identified in the 1500s and defined as a unique clinicopathologic entity in 1809. Until the mid-1900s, knowledge advanced sporadically, with technological developments of ophthalmoscopy and light microscopy, and with the introduction of surgical enucleation, chemotherapy, and radiation therapy. During the last 50 years, research and treatment have progressed at an unprecedented rate owing to innovations in molecular biology and the development of targeted therapies. During this time period, the retinoblastoma gene was discovered; techniques for genetic testing for retinoblastoma were developed; and plaque brachytherapy, chemoreduction, intra-arterial chemotherapy, and intraocular injections of chemotherapeutic agents were successfully introduced.
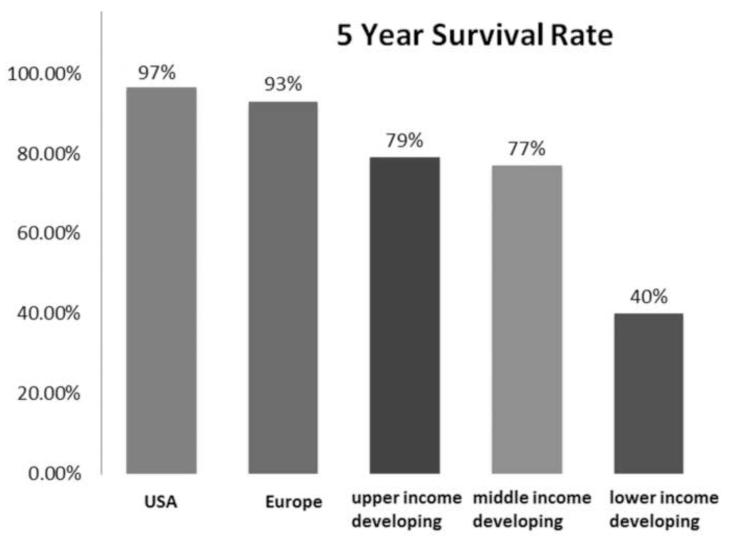
Conclusions: Nearly all patients with retinoblastoma in developed countries can now be cured of their primary cancer--a remarkable achievement for a childhood cancer that once was uniformly fatal. Much of this success is owed to deciphering the role of the Rb gene, and the benefits of targeted therapies, such as chemoreduction with consolidation as well as intra-arterial and intravitreal chemotherapies. Going forward, the main challenge will be ensuring that access to care is available for all children, particularly those in developing countries.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
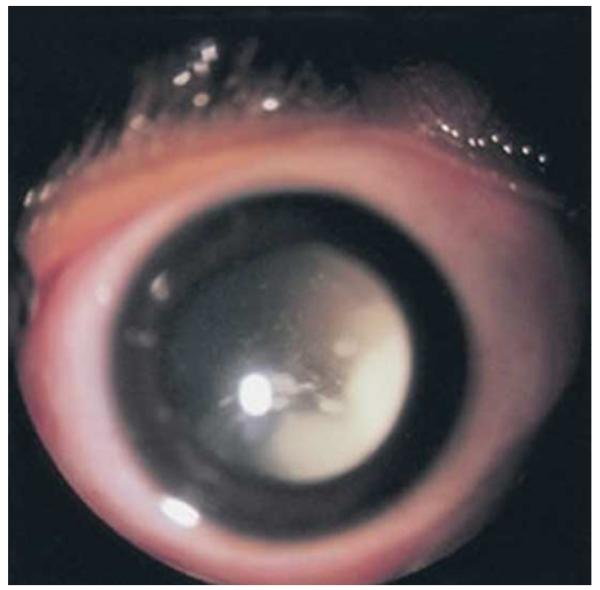
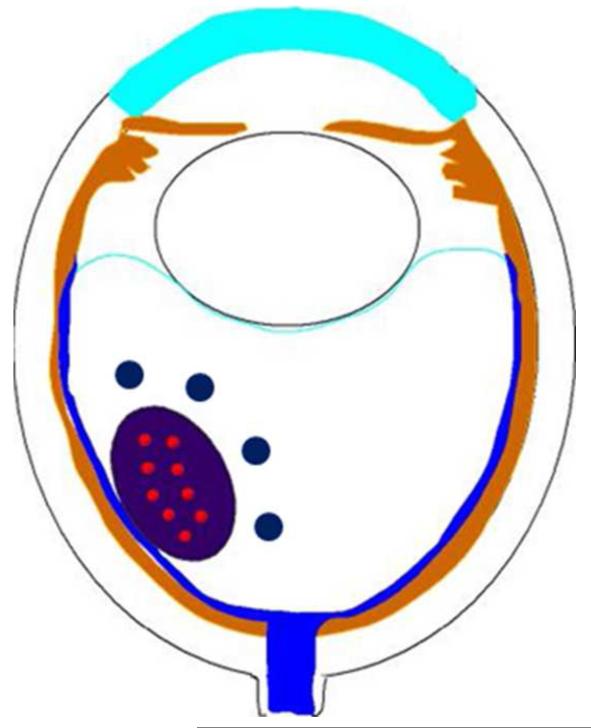
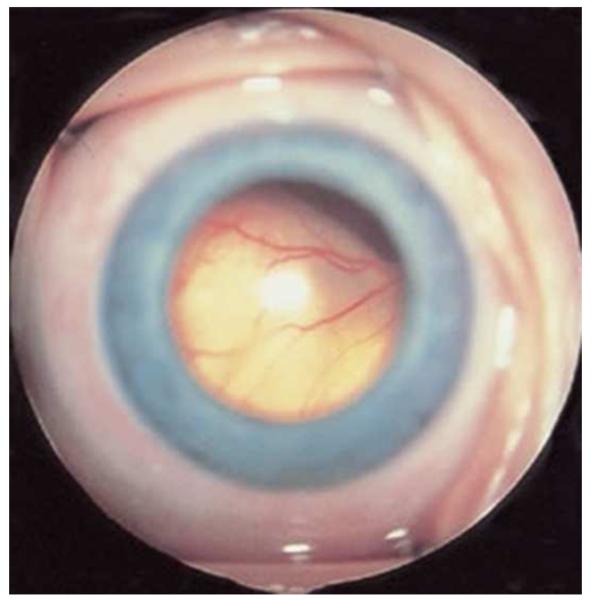
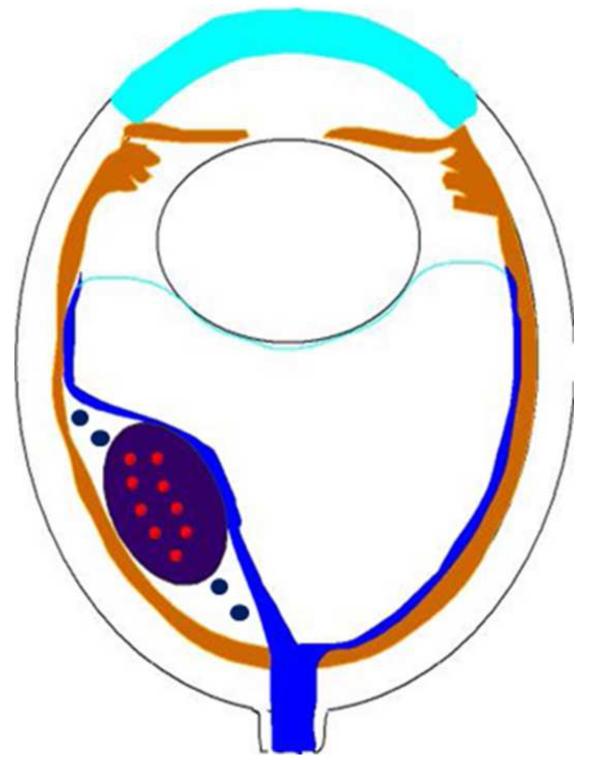
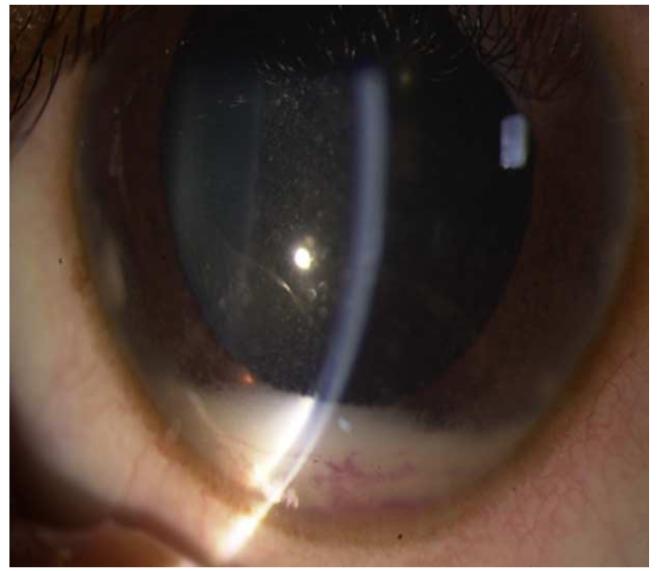
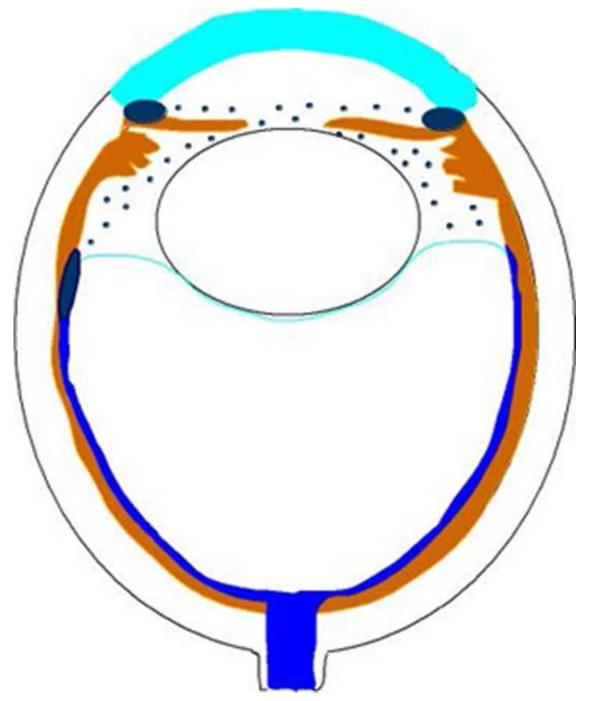
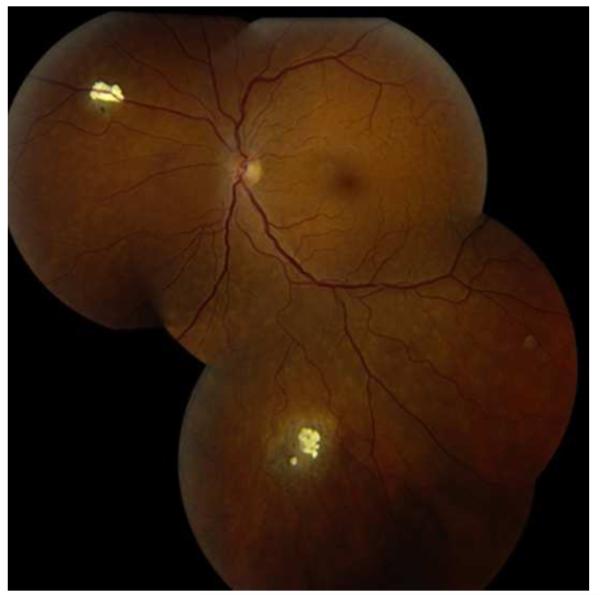
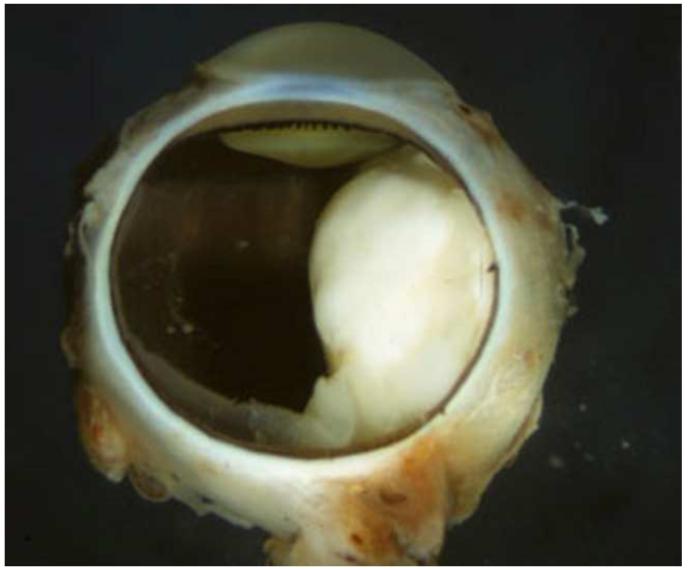
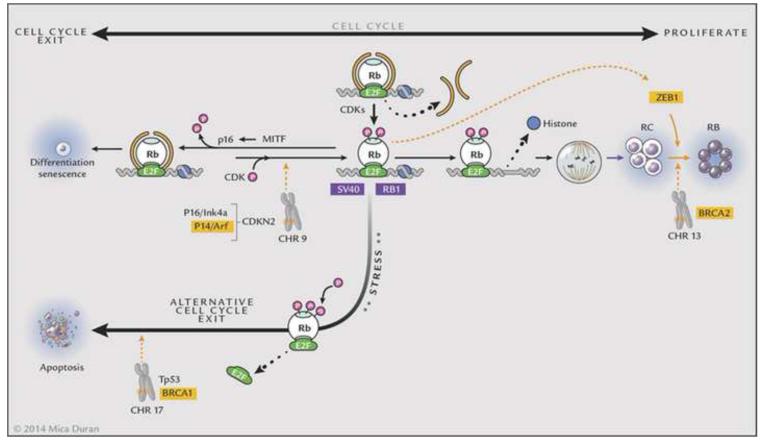
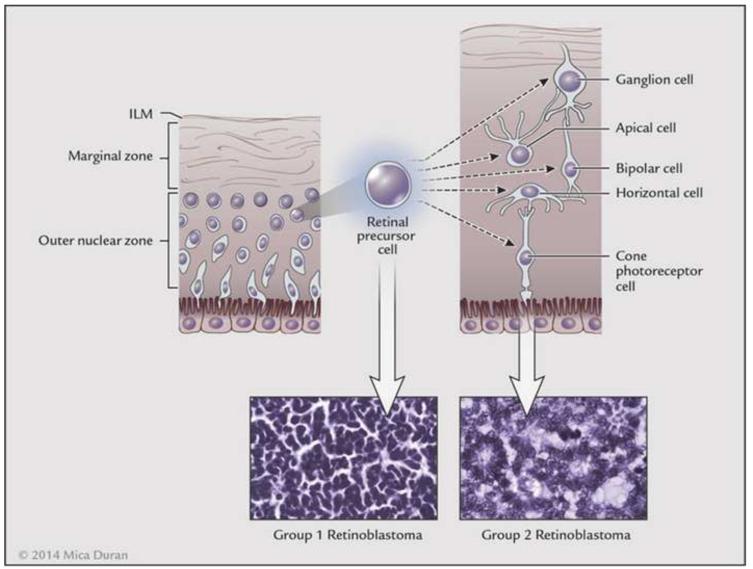
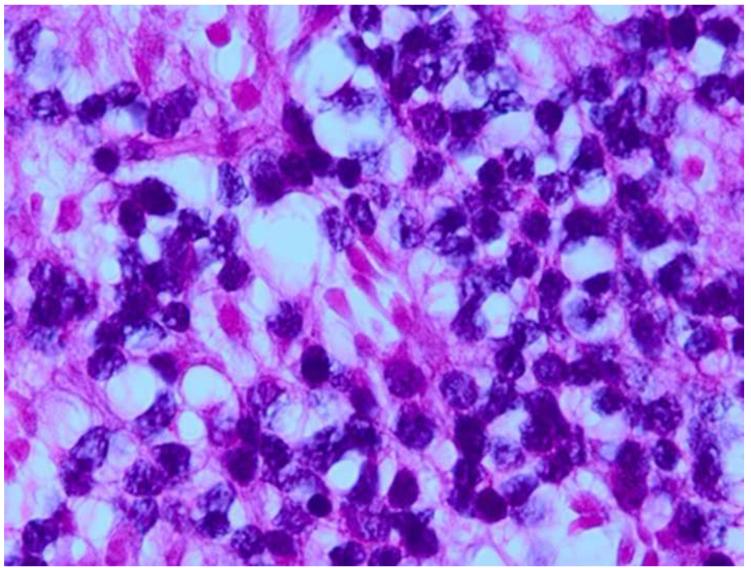
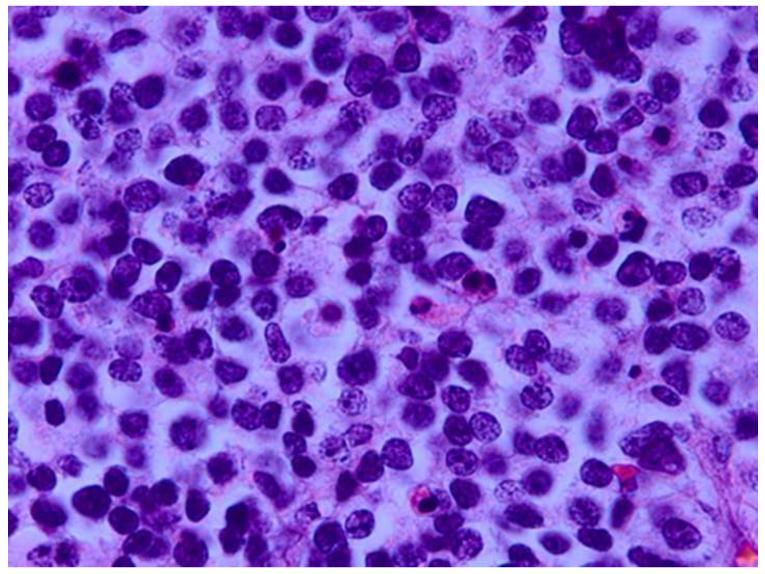
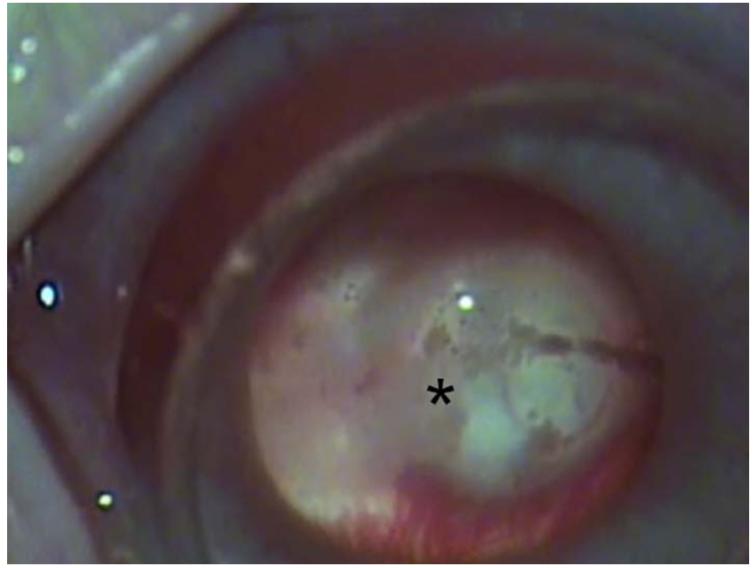
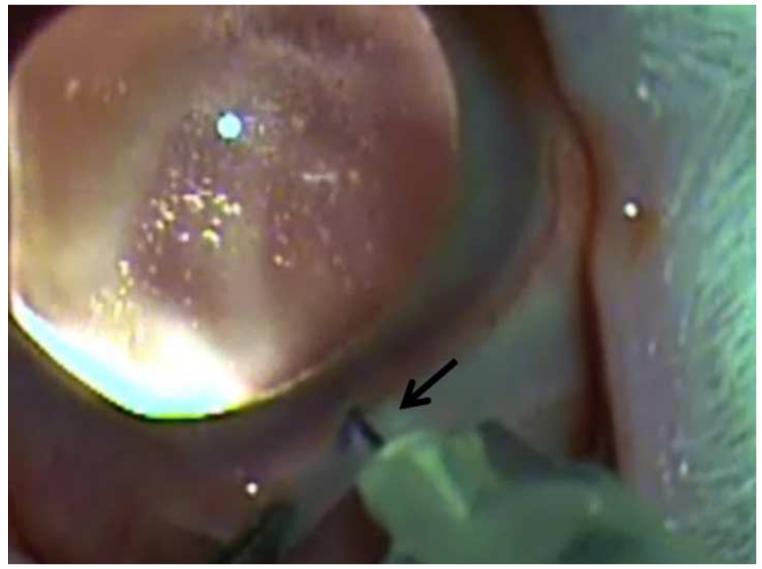
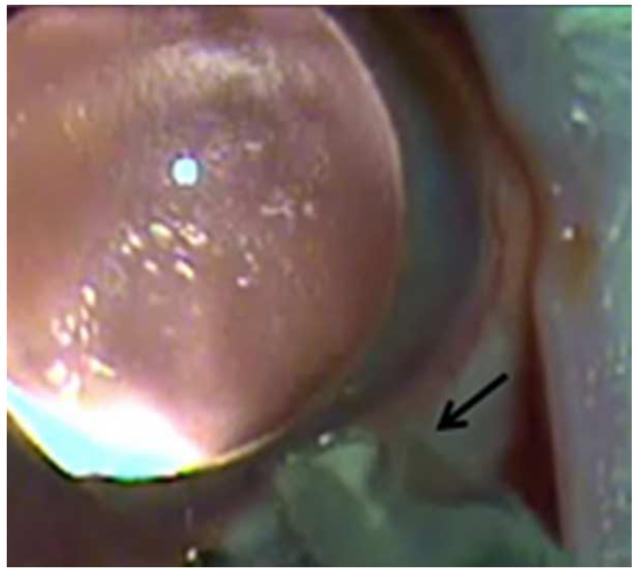
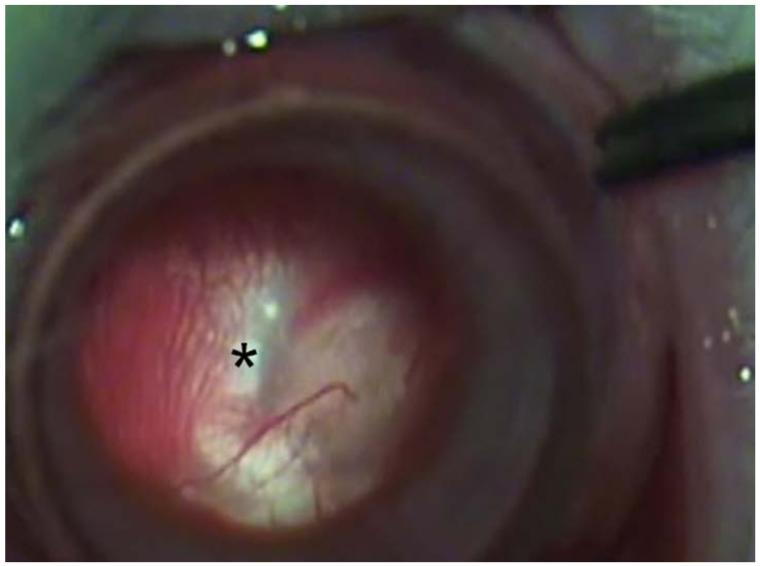
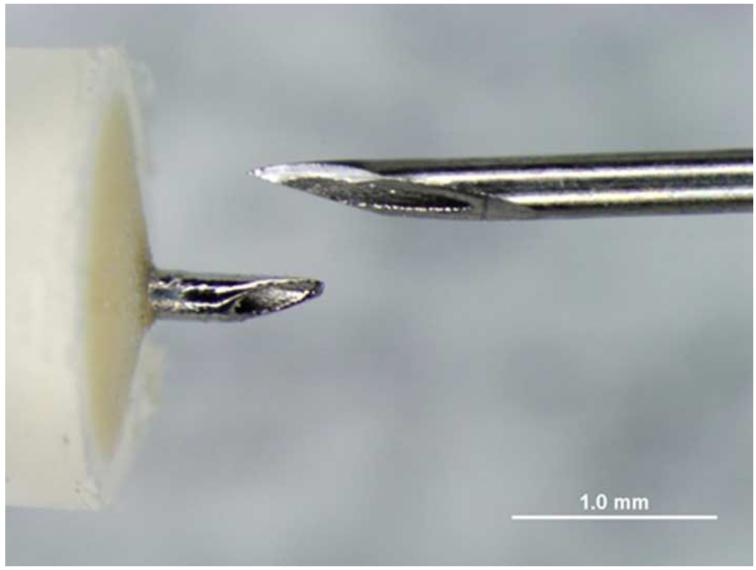
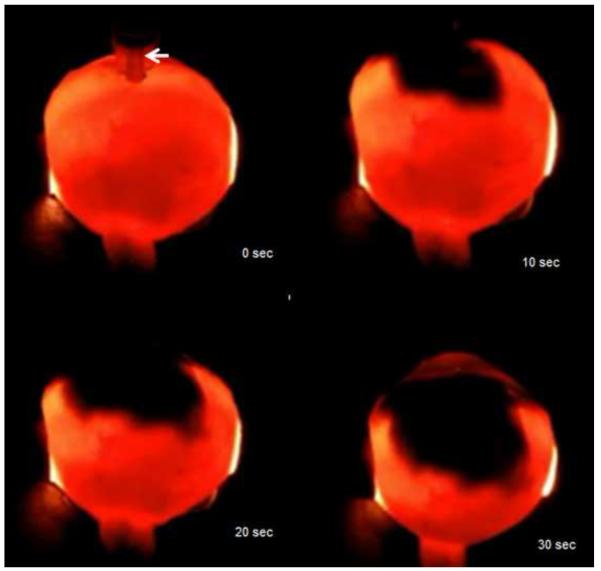
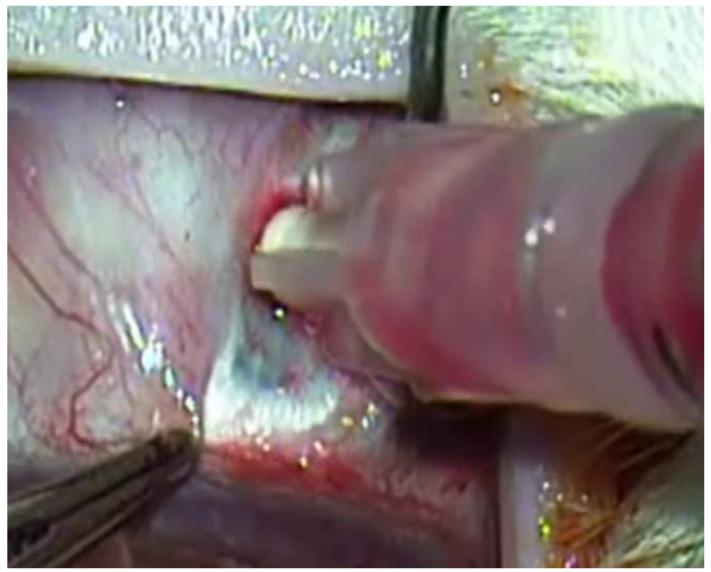
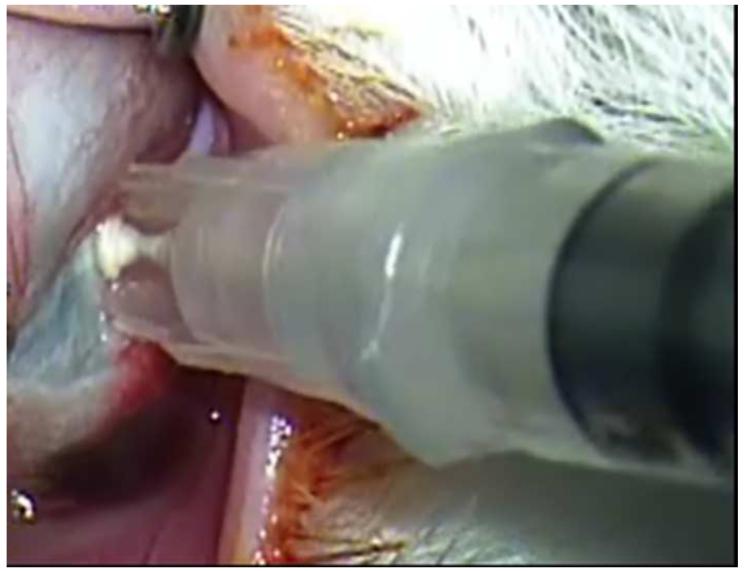
References
-
- Liesegang TJ, Hoskins HD, Jensen AD. The significance of the Edward Jackson lecture. Am J Ophthalmol. 2005;139(3):530–532. - PubMed
-
- Dunphy EB. The story of retinoblastoma. The XX Edward Jackson Memorial Lecture. Am J Ophthalmol. 1964;58(4):539–552. - PubMed
-
- Albert DM. Historic review of retinoblastoma. Ophthalmology. 1987;94(6):654–662. - PubMed
-
- Pawius P. Observationes anatomical. 1657;16:336. Cited by Bartolini, Bartolini Hafniae.
-
- Wardrop J. Observations on the Fungus Haematodes or Soft Cancer. George Ramsay and Co; Edinburgh: 2013. p. 1809.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
