

The Alu-rich genomic architecture of SPAST predisposes to diverse and functionally distinct disease-associated CNV alleles
- PMID: 25065914
- PMCID: PMC4129405
- DOI: 10.1016/j.ajhg.2014.06.014
The Alu-rich genomic architecture of SPAST predisposes to diverse and functionally distinct disease-associated CNV alleles
Abstract
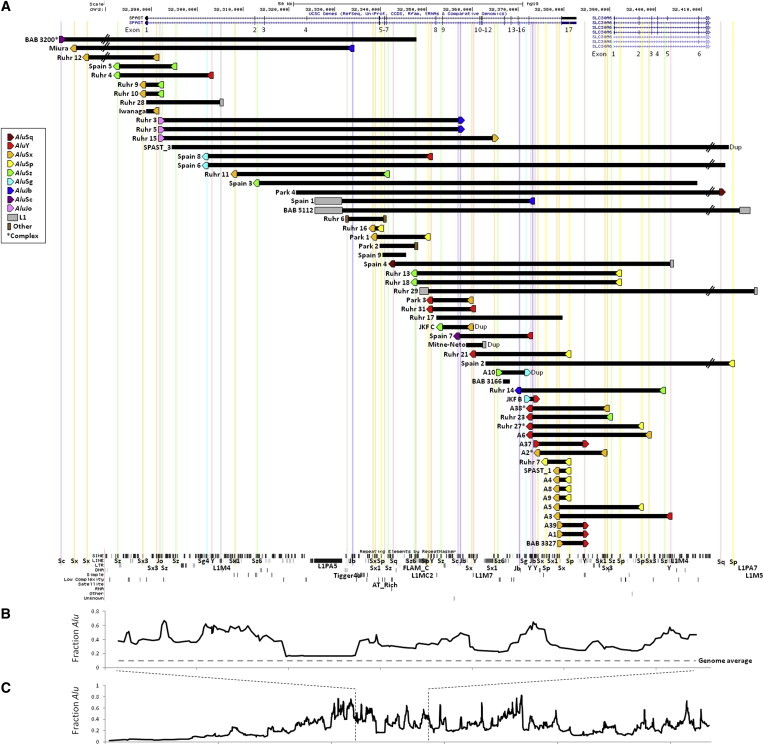
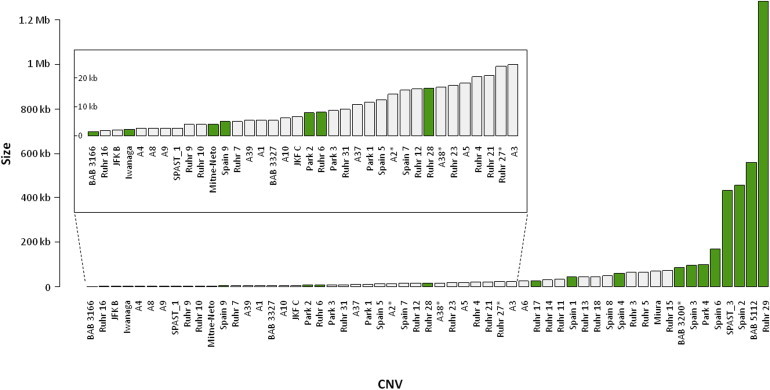
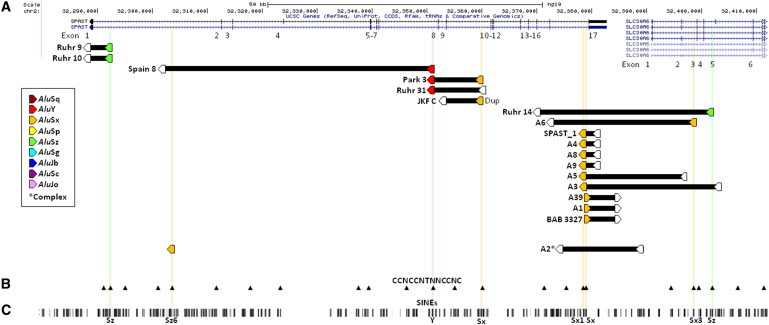
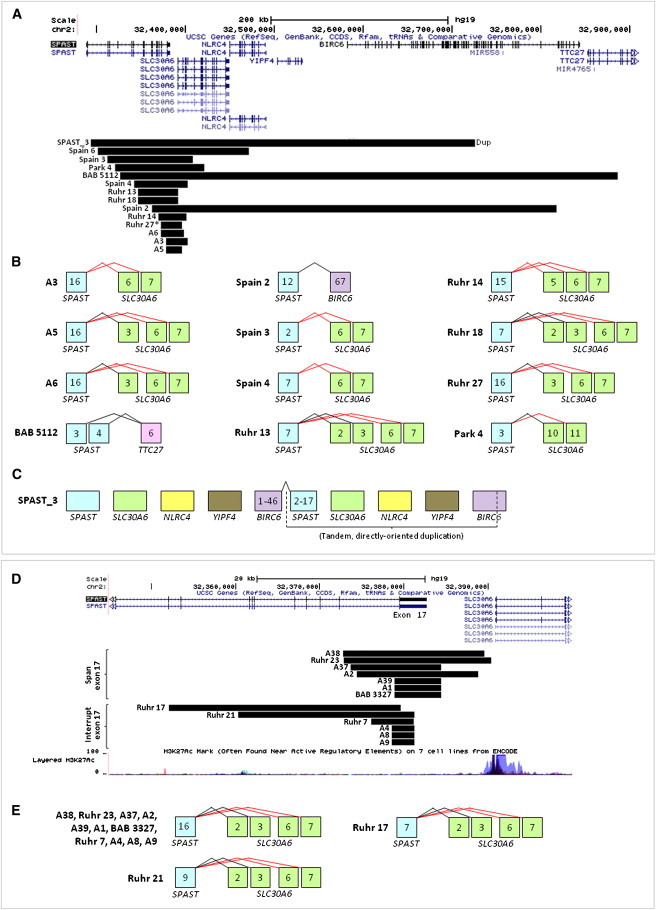
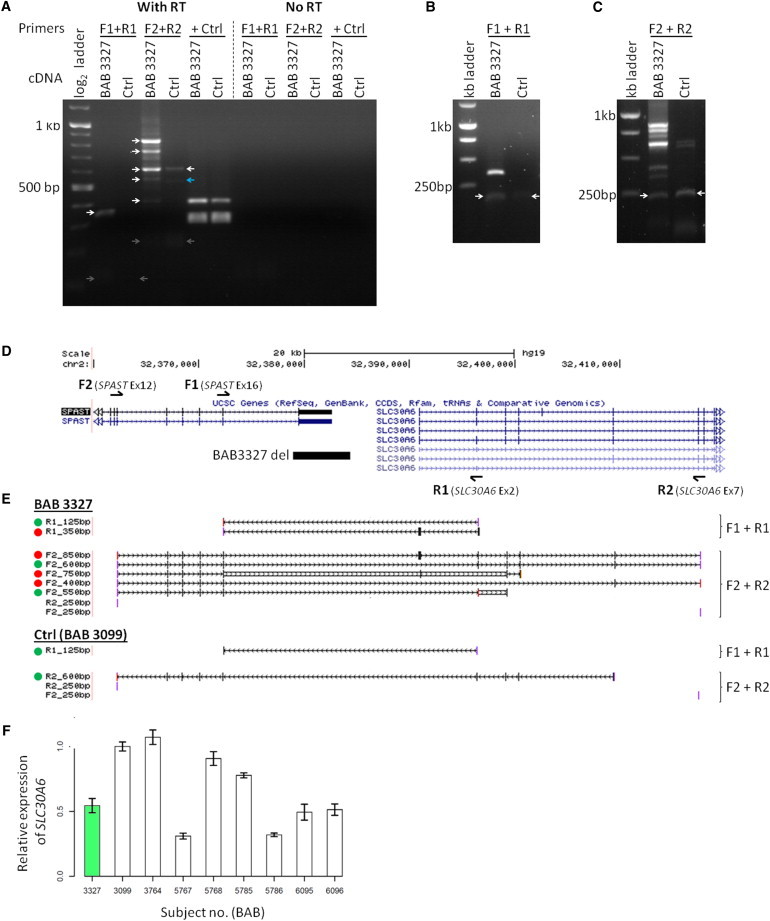
Intragenic copy-number variants (CNVs) contribute to the allelic spectrum of both Mendelian and complex disorders. Although pathogenic deletions and duplications in SPAST (mutations in which cause autosomal-dominant spastic paraplegia 4 [SPG4]) have been described, their origins and molecular consequences remain obscure. We mapped breakpoint junctions of 54 SPAST CNVs at nucleotide resolution. Diverse combinations of exons are deleted or duplicated, highlighting the importance of particular exons for spastin function. Of the 54 CNVs, 38 (70%) appear to be mediated by an Alu-based mechanism, suggesting that the Alu-rich genomic architecture of SPAST renders this locus susceptible to various genome rearrangements. Analysis of breakpoint Alus further informs a model of Alu-mediated CNV formation characterized by small CNV size and potential involvement of mechanisms other than homologous recombination. Twelve deletions (22%) overlap part of SPAST and a portion of a nearby, directly oriented gene, predicting novel chimeric genes in these subjects' genomes. cDNA from a subject with a SPAST final exon deletion contained multiple SPAST:SLC30A6 fusion transcripts, indicating that SPAST CNVs can have transcriptional effects beyond the gene itself. SLC30A6 has been implicated in Alzheimer disease, so these fusion gene data could explain a report of spastic paraplegia and dementia cosegregating in a family with deletion of the final exon of SPAST. Our findings provide evidence that the Alu genomic architecture of SPAST predisposes to diverse CNV alleles with distinct transcriptional--and possibly phenotypic--consequences. Moreover, we provide further mechanistic insights into Alu-mediated copy-number change that are extendable to other loci.
Copyright © 2014 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Wiszniewski W., Hunter J.V., Hanchard N.A., Willer J.R., Shaw C., Tian Q., Illner A., Wang X., Cheung S.W., Patel A. TM4SF20 ancestral deletion and susceptibility to a pediatric disorder of early language delay and cerebral white matter hyperintensities. Am. J. Hum. Genet. 2013;93:197–210. - PMC - PubMed
-
- Benito-Sanz S., Royo J.L., Barroso E., Paumard-Hernández B., Barreda-Bonis A.C., Liu P., Gracía R., Lupski J.R., Campos-Barros Á., Gómez-Skarmeta J.L., Heath K.E. Identification of the first recurrent PAR1 deletion in Léri-Weill dyschondrosteosis and idiopathic short stature reveals the presence of a novel SHOX enhancer. J. Med. Genet. 2012;49:442–450. - PubMed
-
- Loureiro J.L., Brandão E., Ruano L., Brandão A.F., Lopes A.M., Thieleke-Matos C., Miller-Fleming L., Cruz V.T., Barbosa M., Silveira I. Autosomal dominant spastic paraplegias: a review of 89 families resulting from a portuguese survey. JAMA Neurol. 2013;70:481–487. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- R01NS0679700/NS/NINDS NIH HHS/United States
- U54 HD083092/HD/NICHD NIH HHS/United States
- UM1 HG006542/HG/NHGRI NIH HHS/United States
- T32GM007330/GM/NIGMS NIH HHS/United States
- R01 NS069700/NS/NINDS NIH HHS/United States
- T32 NS043124/NS/NINDS NIH HHS/United States
- F31 NS083159/NS/NINDS NIH HHS/United States
- U54 HG006542/HG/NHGRI NIH HHS/United States
- I01 CX000344/CX/CSRD VA/United States
- R01NS058529/NS/NINDS NIH HHS/United States
- U54HG006542/HG/NHGRI NIH HHS/United States
- T32 GM007330/GM/NIGMS NIH HHS/United States
- R01 NS058529/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
