

Compartmentalized acyl-CoA metabolism in skeletal muscle regulates systemic glucose homeostasis
- PMID: 25071025
- PMCID: PMC4274800
- DOI: 10.2337/db13-1070
Compartmentalized acyl-CoA metabolism in skeletal muscle regulates systemic glucose homeostasis
Abstract
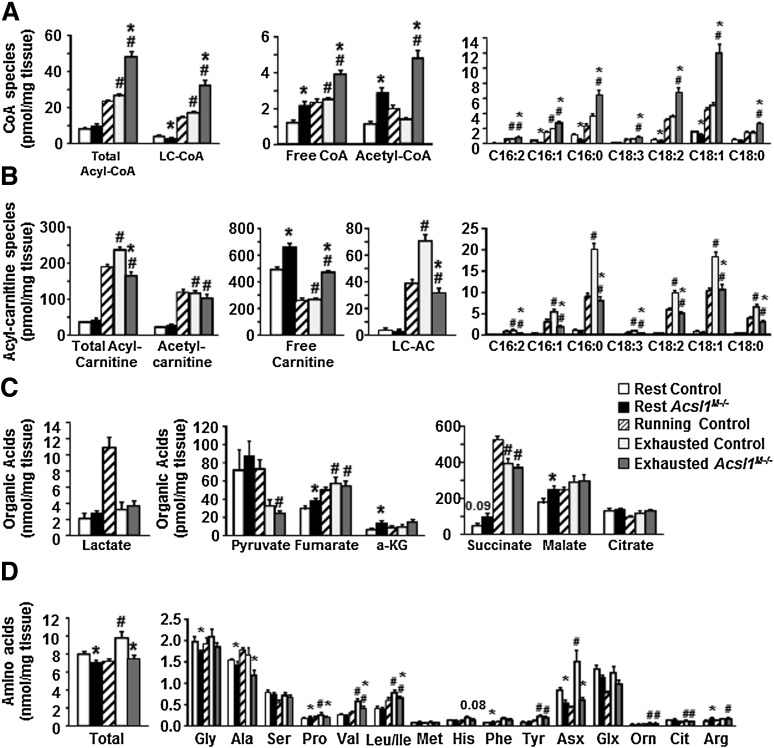
The impaired capacity of skeletal muscle to switch between the oxidation of fatty acid (FA) and glucose is linked to disordered metabolic homeostasis. To understand how muscle FA oxidation affects systemic glucose, we studied mice with a skeletal muscle-specific deficiency of long-chain acyl-CoA synthetase (ACSL)1. ACSL1 deficiency caused a 91% loss of ACSL-specific activity and a 60-85% decrease in muscle FA oxidation. Acsl1(M-/-) mice were more insulin sensitive, and, during an overnight fast, their respiratory exchange ratio was higher, indicating greater glucose use. During endurance exercise, Acsl1(M-/-) mice ran only 48% as far as controls. At the time that Acsl1(M-/-) mice were exhausted but control mice continued to run, liver and muscle glycogen and triacylglycerol stores were similar in both genotypes; however, plasma glucose concentrations in Acsl1(M-/-) mice were ∼40 mg/dL, whereas glucose concentrations in controls were ∼90 mg/dL. Excess use of glucose and the likely use of amino acids for fuel within muscle depleted glucose reserves and diminished substrate availability for hepatic gluconeogenesis. Surprisingly, the content of muscle acyl-CoA at exhaustion was markedly elevated, indicating that acyl-CoAs synthesized by other ACSL isoforms were not available for β-oxidation. This compartmentalization of acyl-CoAs resulted in both an excessive glucose requirement and severely compromised systemic glucose homeostasis.
© 2015 by the American Diabetes Association. Readers may use this article as long as the work is properly cited, the use is educational and not for profit, and the work is not altered.
Figures






References
-
- Kelley DE, Mandarino LJ. Fuel selection in human skeletal muscle in insulin resistance: a reexamination. Diabetes 2000;49:677–683 - PubMed
-
- Thyfault JP, Rector RS, Noland RC. Metabolic inflexibility in skeletal muscle: a prelude to the cardiometabolic syndrome? J Cardiometab Syndr 2006;1:184–189 - PubMed
-
- Watt MJ, Hoy AJ. Lipid metabolism in skeletal muscle: generation of adaptive and maladaptive intracellular signals for cellular function. Am J Physiol Endocrinol Metab 2012;302:E1315–E1328 - PubMed
-
- Ukropcova B, Sereda O, de Jonge L, et al. . Family history of diabetes links impaired substrate switching and reduced mitochondrial content in skeletal muscle. Diabetes 2007;56:720–727 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
