

CRTC1/MAML2 gain-of-function interactions with MYC create a gene signature predictive of cancers with CREB-MYC involvement
- PMID: 25071166
- PMCID: PMC4136562
- DOI: 10.1073/pnas.1319176111
CRTC1/MAML2 gain-of-function interactions with MYC create a gene signature predictive of cancers with CREB-MYC involvement
Abstract
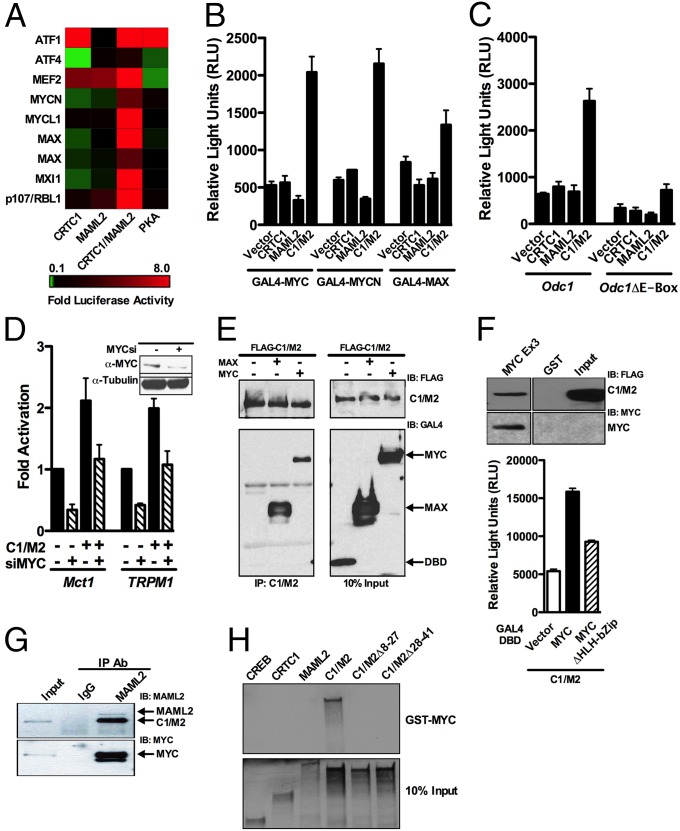
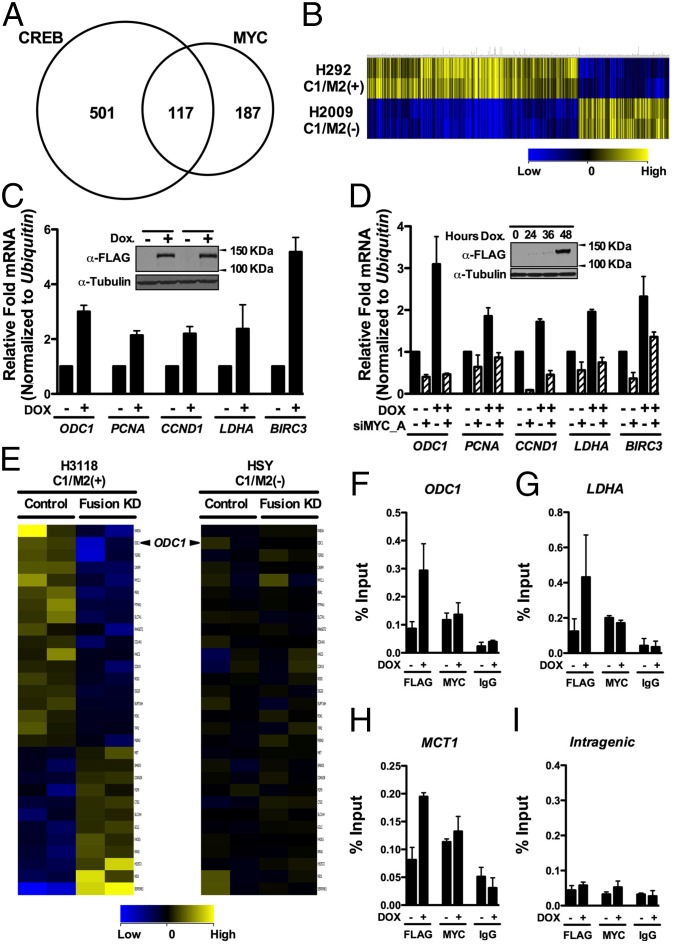
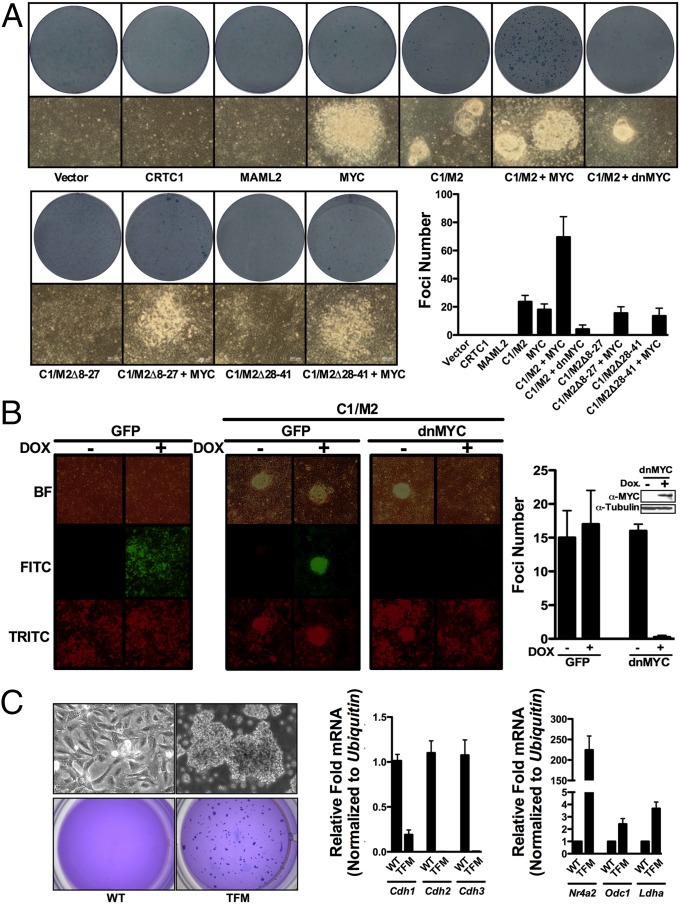
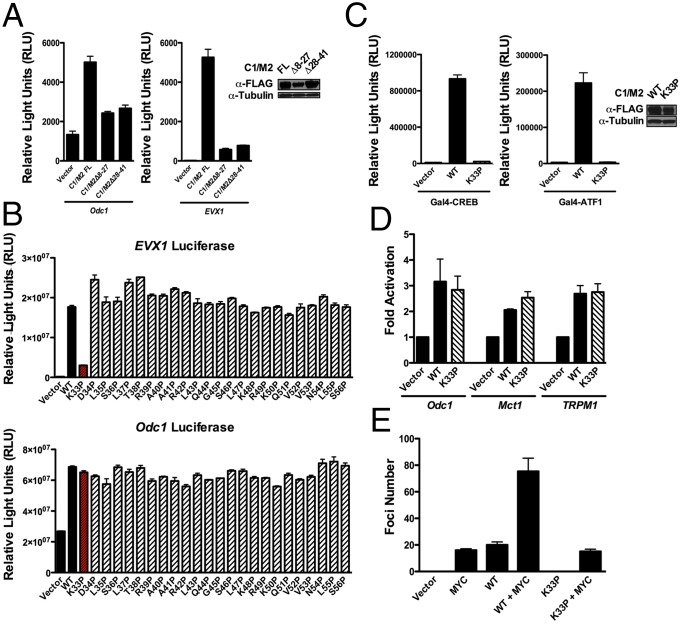
Chimeric oncoproteins created by chromosomal translocations are among the most common genetic mutations associated with tumorigenesis. Malignant mucoepidermoid salivary gland tumors, as well as a growing number of solid epithelial-derived tumors, can arise from a recurrent t (11, 19)(q21;p13.1) translocation that generates an unusual chimeric cAMP response element binding protein (CREB)-regulated transcriptional coactivator 1 (CRTC1)/mastermind-like 2 (MAML2) (C1/M2) oncoprotein comprised of two transcriptional coactivators, the CRTC1 and the NOTCH/RBPJ coactivator MAML2. Accordingly, the C1/M2 oncoprotein induces aberrant expression of CREB and NOTCH target genes. Surprisingly, here we report a gain-of-function activity of the C1/M2 oncoprotein that directs its interactions with myelocytomatosis oncogene (MYC) proteins and the activation of MYC transcription targets, including those involved in cell growth and metabolism, survival, and tumorigenesis. These results were validated in human mucoepidermoid tumor cells that harbor the t (11, 19)(q21;p13.1) translocation and express the C1/M2 oncoprotein. Notably, the C1/M2-MYC interaction is necessary for C1/M2-driven cell transformation, and the C1/M2 transcriptional signature predicts other human malignancies having combined involvement of MYC and CREB. These findings suggest that such gain-of-function properties may also be manifest in other oncoprotein fusions found in human cancer and that agents targeting the C1/M2-MYC interface represent an attractive strategy for the development of effective and safe anticancer therapeutics in tumors harboring the t (11, 19) translocation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. - PubMed
-
- Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144(5):646–674. - PubMed
-
- Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10(8):789–799. - PubMed
-
- Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7(4):233–245. - PubMed
-
- Rabbitts TH. Commonality but diversity in cancer gene fusions. Cell. 2009;137(3):391–395. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
