

Ubiquitin pathways in neurodegenerative disease
- PMID: 25071440
- PMCID: PMC4085722
- DOI: 10.3389/fnmol.2014.00063
Ubiquitin pathways in neurodegenerative disease
Abstract
Control of proper protein synthesis, function, and turnover is essential for the health of all cells. In neurons these demands take on the additional importance of supporting and regulating the highly dynamic connections between neurons that are necessary for cognitive function, learning, and memory. Regulating multiple unique synaptic protein environments within a single neuron while maintaining cell health requires the highly regulated processes of ubiquitination and degradation of ubiquitinated proteins through the proteasome. In this review, we examine the effects of dysregulated ubiquitination and protein clearance on the handling of disease-associated proteins and neuronal health in the most common neurodegenerative diseases.
Keywords: Alzheimer's disease; Amyotrophic Lateral Sclerosis; Huntington's disease; Parkinson disease; neurodegenerative diseases; proteasome; protein quality control; ubiquitin.
Figures
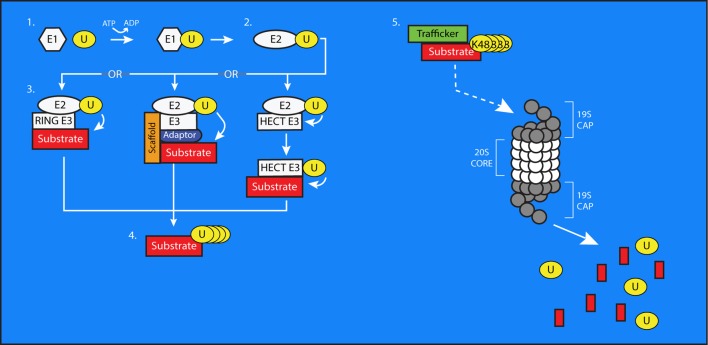

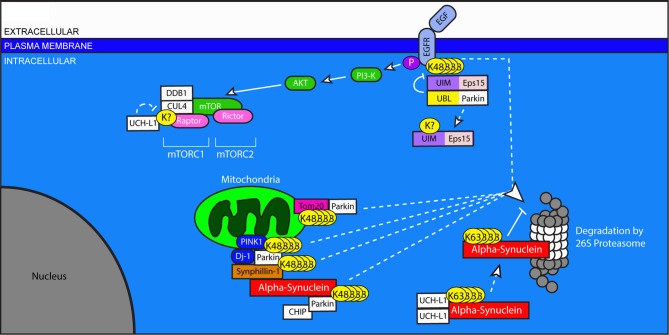


References
-
- Adachi H., Waza M., Tokui K., Katsuno M., Minamiyama M., Tanaka F., et al. (2007). CHIP overexpression reduces mutant androgen receptor protein and ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model. J. Neurosci. 27, 5115–5126 10.1523/JNEUROSCI.1242-07.2007 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources