

Dysfunctional mitochondrial Ca(2+) handling in mutant SOD1 mouse models of fALS: integration of findings from motor neuron somata and motor terminals
- PMID: 25071445
- PMCID: PMC4085874
- DOI: 10.3389/fncel.2014.00184
Dysfunctional mitochondrial Ca(2+) handling in mutant SOD1 mouse models of fALS: integration of findings from motor neuron somata and motor terminals
Abstract
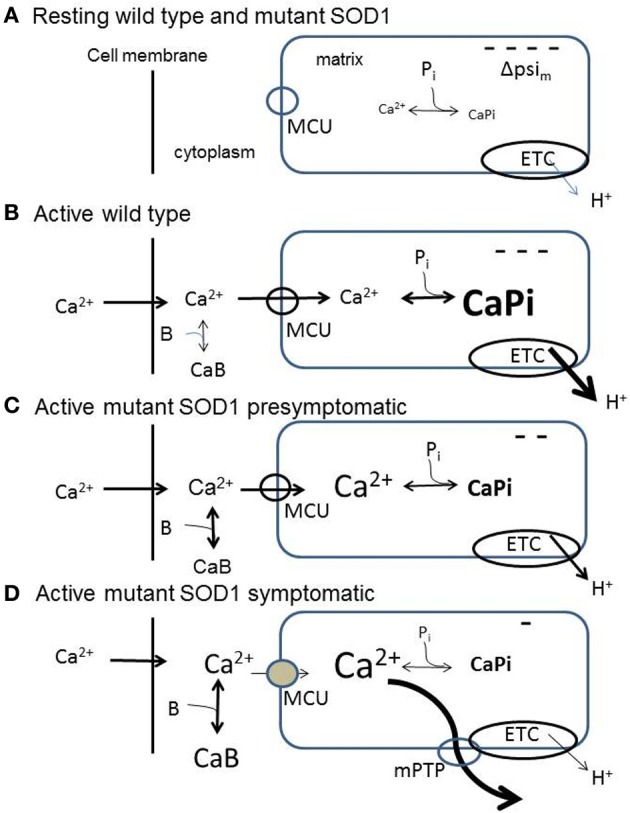
Abundant evidence indicates that mitochondrial dysfunction and Ca(2+) dysregulation contribute to the muscle denervation and motor neuron death that occur in mouse models of familial amyotrophic lateral sclerosis (fALS). This perspective considers measurements of mitochondrial function and Ca(2+) handling made in both motor neuron somata and motor nerve terminals of SOD1-G93A mice at different disease stages. These complementary studies are integrated into a model of how mitochondrial dysfunction disrupts handling of stimulation-induced Ca(2+) loads in presymptomatic and end-stages of this disease. Also considered are possible mechanisms underlying the findings that some treatments that preserve motor neuron somata fail to postpone degeneration of motor axons and terminals.
Keywords: Ca2+ regulation; mitochondria; motor nerve terminal; motor neuron; mutant SOD1 models of fALS.
Figures

Similar articles
-
Repetitive nerve stimulation transiently opens the mitochondrial permeability transition pore in motor nerve terminals of symptomatic mutant SOD1 mice.Neurobiol Dis. 2011 Jun;42(3):381-90. doi: 10.1016/j.nbd.2011.01.031. Epub 2011 Feb 18. Neurobiol Dis. 2011. PMID: 21310237 Free PMC article.
-
Early vulnerability to ischemia/reperfusion injury in motor terminals innervating fast muscles of SOD1-G93A mice.Exp Neurol. 2007 Mar;204(1):411-20. doi: 10.1016/j.expneurol.2006.12.021. Epub 2007 Jan 4. Exp Neurol. 2007. PMID: 17292357 Free PMC article.
-
The Psi(m) depolarization that accompanies mitochondrial Ca2+ uptake is greater in mutant SOD1 than in wild-type mouse motor terminals.Proc Natl Acad Sci U S A. 2009 Feb 10;106(6):2007-11. doi: 10.1073/pnas.0810934106. Epub 2009 Jan 27. Proc Natl Acad Sci U S A. 2009. PMID: 19174508 Free PMC article.
-
Oxidative stress, mutant SOD1, and neurofilament pathology in transgenic mouse models of human motor neuron disease.Lab Invest. 1997 Apr;76(4):441-56. Lab Invest. 1997. PMID: 9111507 Review.
-
Transgenic mice with human mutant genes causing Parkinson's disease and amyotrophic lateral sclerosis provide common insight into mechanisms of motor neuron selective vulnerability to degeneration.Rev Neurosci. 2007;18(2):115-36. doi: 10.1515/revneuro.2007.18.2.115. Rev Neurosci. 2007. PMID: 17593875 Review.
Cited by
-
Structure and Function of the Mammalian Neuromuscular Junction.Compr Physiol. 2022 Aug 11;12(4):3731-3766. doi: 10.1002/cphy.c210022. Compr Physiol. 2022. PMID: 35950651 Free PMC article.
-
Spinal Cord Injury Model Mitochondria Connect Altered Function with Defects of Mitochondrion Morphology: an Ultrastructural Study.Mol Neurobiol. 2024 Apr;61(4):2241-2248. doi: 10.1007/s12035-023-03710-3. Epub 2023 Oct 23. Mol Neurobiol. 2024. PMID: 37870678
-
Functional Importance of Transient Receptor Potential (TRP) Channels in Neurological Disorders.Front Cell Dev Biol. 2021 Mar 4;9:611773. doi: 10.3389/fcell.2021.611773. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 33748103 Free PMC article. Review.
-
Homocysteine aggravates ROS-induced depression of transmitter release from motor nerve terminals: potential mechanism of peripheral impairment in motor neuron diseases associated with hyperhomocysteinemia.Front Cell Neurosci. 2015 Oct 6;9:391. doi: 10.3389/fncel.2015.00391. eCollection 2015. Front Cell Neurosci. 2015. PMID: 26500495 Free PMC article.
-
Mitochondrial Calcium: Effects of Its Imbalance in Disease.Antioxidants (Basel). 2022 Apr 20;11(5):801. doi: 10.3390/antiox11050801. Antioxidants (Basel). 2022. PMID: 35624667 Free PMC article. Review.
References
-
- Bros-Facer V., Krull D., Taylor A., Dick J. R., Bates S. A., Cleveland M. S., et al. (2014). Treatment with an antibody directed against Nogo-A delays disease progression in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Hum. Mol. Genet. [Epub ahead of print]. 10.1093/hmg/ddu136 - DOI - PubMed
-
- Burke R. E. (2004). The structure and function of motor units, in Myology, 3rd Edn., eds Engel A. G., Franzini-Armstrong C. (New York, NY: McGraw-Hill; ), 104–118
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous