

Polarized axonal surface expression of neuronal KCNQ potassium channels is regulated by calmodulin interaction with KCNQ2 subunit
- PMID: 25077630
- PMCID: PMC4117524
- DOI: 10.1371/journal.pone.0103655
Polarized axonal surface expression of neuronal KCNQ potassium channels is regulated by calmodulin interaction with KCNQ2 subunit
Abstract
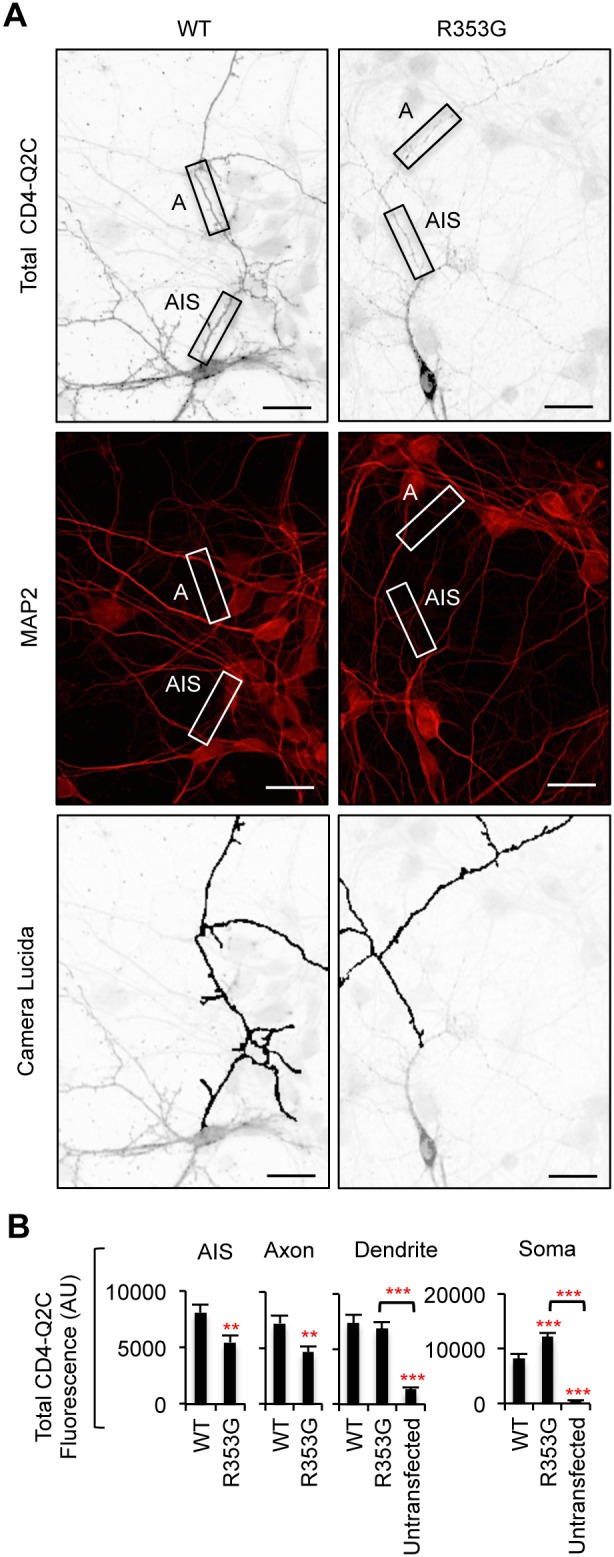
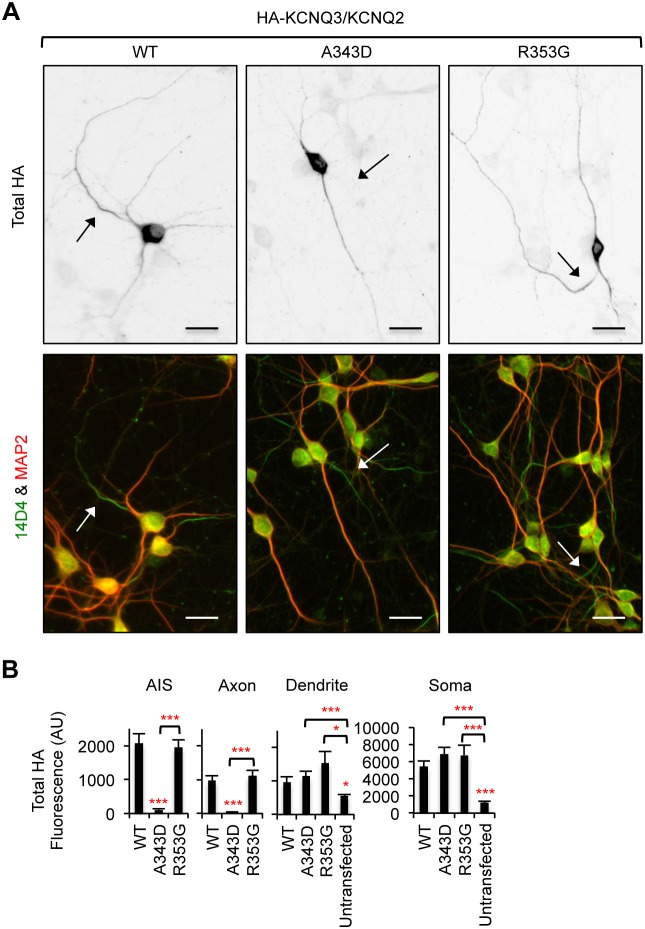
KCNQ potassium channels composed of KCNQ2 and KCNQ3 subunits give rise to the M-current, a slow-activating and non-inactivating voltage-dependent potassium current that limits repetitive firing of action potentials. KCNQ channels are enriched at the surface of axons and axonal initial segments, the sites for action potential generation and modulation. Their enrichment at the axonal surface is impaired by mutations in KCNQ2 carboxy-terminal tail that cause benign familial neonatal convulsion and myokymia, suggesting that their correct surface distribution and density at the axon is crucial for control of neuronal excitability. However, the molecular mechanisms responsible for regulating enrichment of KCNQ channels at the neuronal axon remain elusive. Here, we show that enrichment of KCNQ channels at the axonal surface of dissociated rat hippocampal cultured neurons is regulated by ubiquitous calcium sensor calmodulin. Using immunocytochemistry and the cluster of differentiation 4 (CD4) membrane protein as a trafficking reporter, we demonstrate that fusion of KCNQ2 carboxy-terminal tail is sufficient to target CD4 protein to the axonal surface whereas inhibition of calmodulin binding to KCNQ2 abolishes axonal surface expression of CD4 fusion proteins by retaining them in the endoplasmic reticulum. Disruption of calmodulin binding to KCNQ2 also impairs enrichment of heteromeric KCNQ2/KCNQ3 channels at the axonal surface by blocking their trafficking from the endoplasmic reticulum to the axon. Consistently, hippocampal neuronal excitability is dampened by transient expression of wild-type KCNQ2 but not mutant KCNQ2 deficient in calmodulin binding. Furthermore, coexpression of mutant calmodulin, which can interact with KCNQ2/KCNQ3 channels but not calcium, reduces but does not abolish their enrichment at the axonal surface, suggesting that apo calmodulin but not calcium-bound calmodulin is necessary for their preferential targeting to the axonal surface. These findings collectively reveal calmodulin as a critical player that modulates trafficking and enrichment of KCNQ channels at the neuronal axon.
Conflict of interest statement
Figures











References
-
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, et al. (1998) KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282: 1890–1893. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
