

Extraction and annotation of human mitochondrial genomes from 1000 Genomes Whole Exome Sequencing data
- PMID: 25077682
- PMCID: PMC4083402
- DOI: 10.1186/1471-2164-15-S3-S2
Extraction and annotation of human mitochondrial genomes from 1000 Genomes Whole Exome Sequencing data
Abstract
Background: Whole Exome Sequencing (WES) is one of the most used and cost-effective next generation technologies that allows sequencing of all nuclear exons. Off-target regions may be captured if they present high sequence similarity with baits. Bioinformatics tools have been optimized to retrieve a large amount of WES off-target mitochondrial DNA (mtDNA), by exploiting the aspecificity of probes, partially overlapping to Nuclear mitochondrial Sequences (NumtS). The 1000 Genomes project represents one of the widest resources to extract mtDNA sequences from WES data, considering the large effort the scientific community is undertaking to reconstruct human population history using mtDNA as marker, and the involvement of mtDNA in pathology.
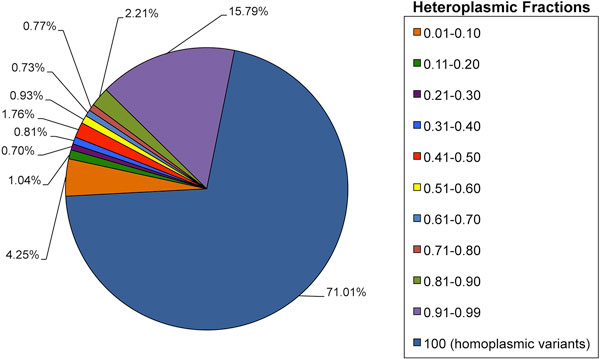
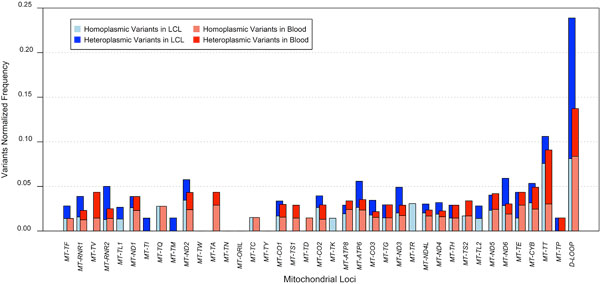
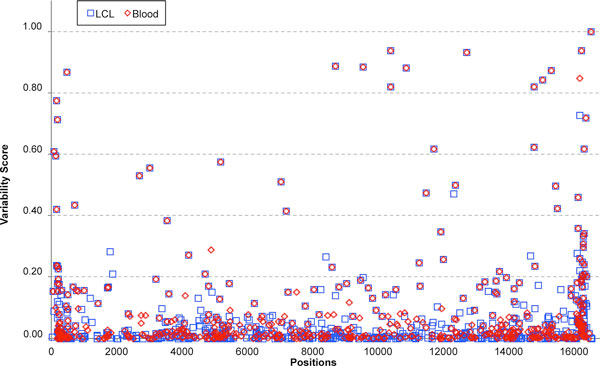
Results: A previously published pipeline aimed at assembling mitochondrial genomes from off-target WES reads and further improved to detect insertions and deletions (indels) and heteroplasmy in a dataset of 1242 samples from the 1000 Genomes project, enabled to obtain a nearly complete mitochondrial genome from 943 samples (76% analyzed exomes). The robustness of our computational strategy was highlighted by the reduction of reads amount recognized as mitochondrial in the original annotation produced by the Consortium, due to NumtS filtering.
Conclusions: To the best of our knowledge, this is likely the most extended population-scale mitochondrial genotyping in humans enriched with the estimation of heteroplasmies.
Figures


References
-
- Tang S, Huang T. Characterization of mitochondrial DNA heteroplasmy using a parallel sequencing system. Biotechniques. 2010;15(4):287–296. - PubMed
-
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;15(11):745–755. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
