

Endoplasmic reticulum stress is chronically activated in chronic pancreatitis
- PMID: 25077966
- PMCID: PMC4183795
- DOI: 10.1074/jbc.M113.528174
Endoplasmic reticulum stress is chronically activated in chronic pancreatitis
Abstract
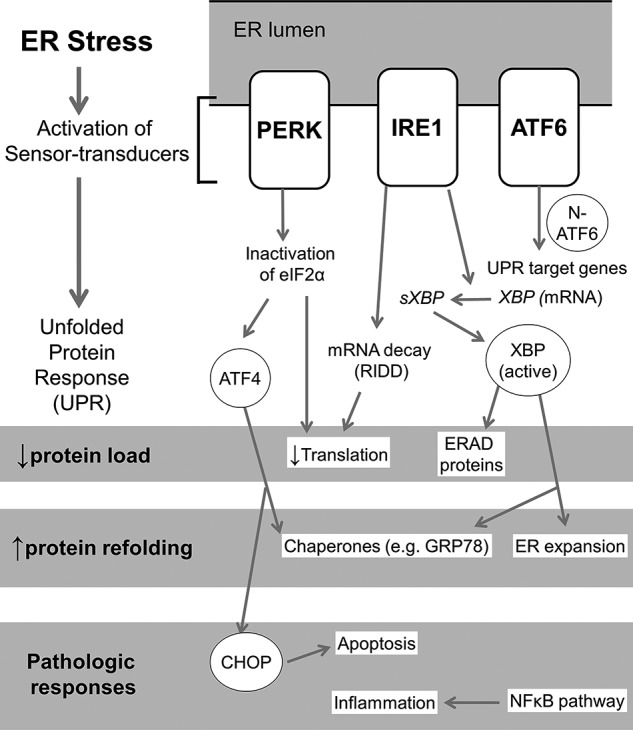
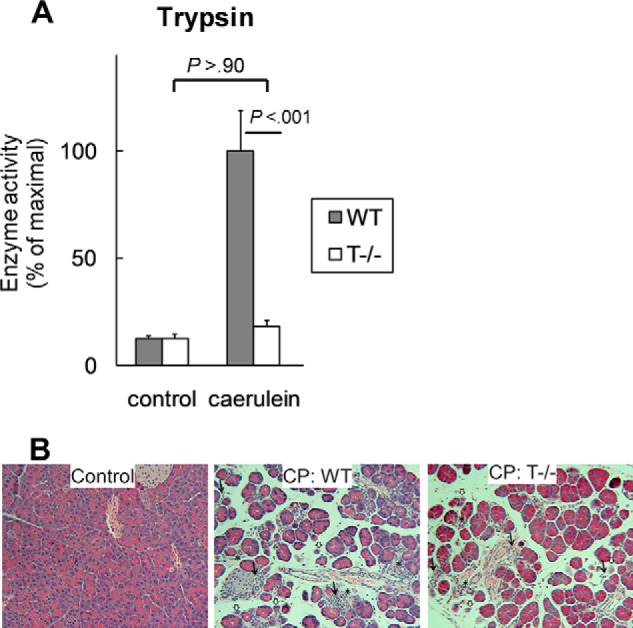
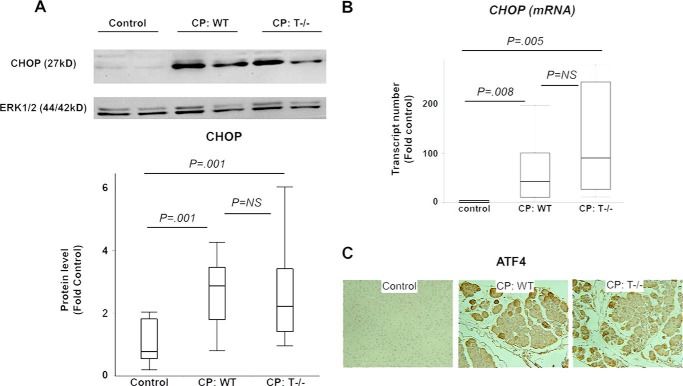
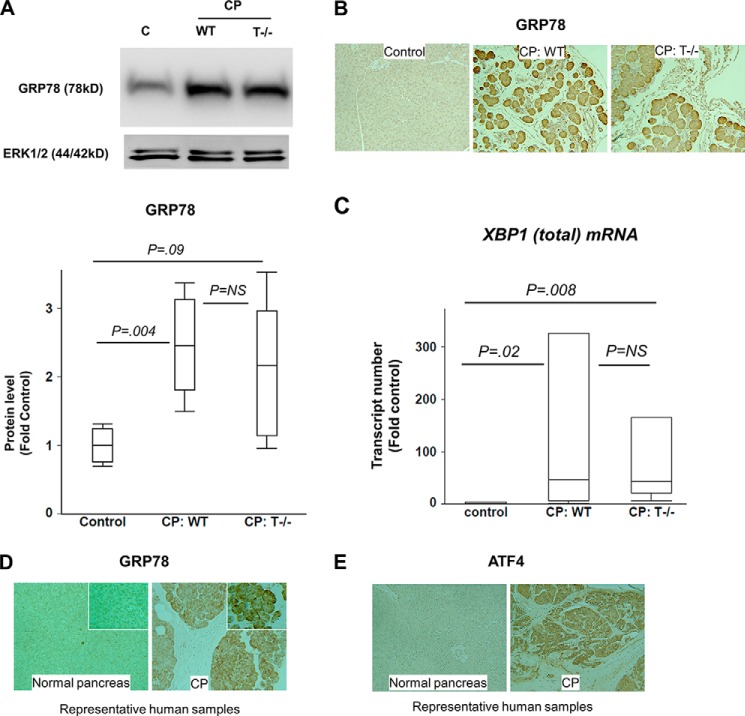
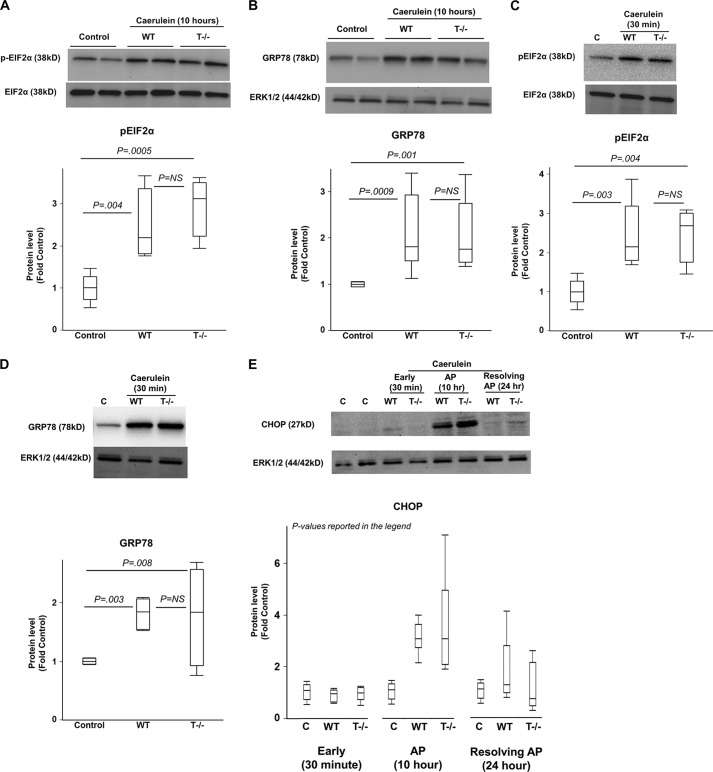
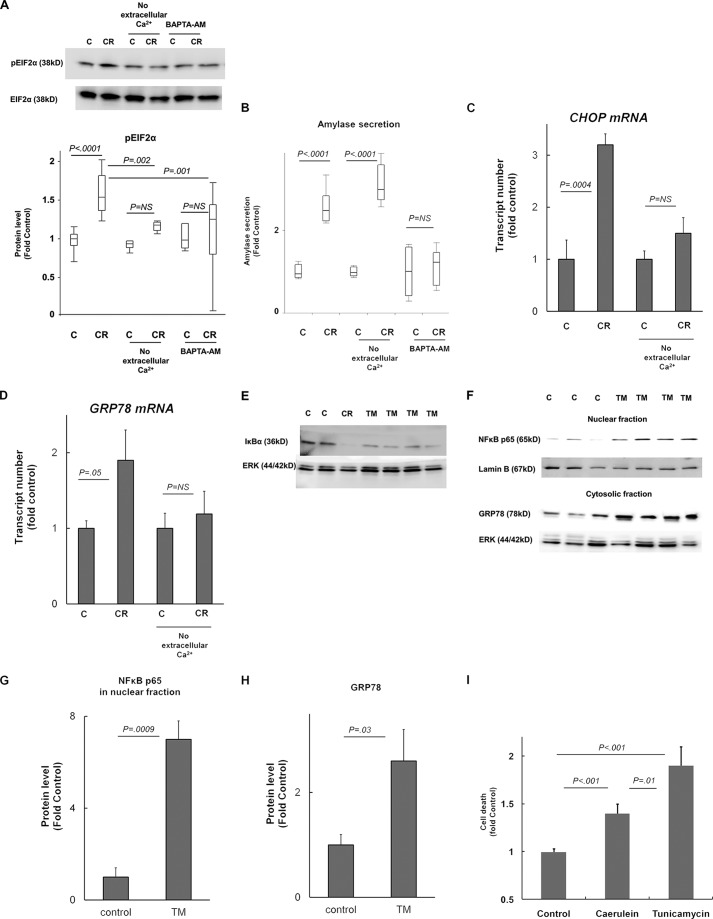
The pathogenesis of chronic pancreatitis (CP) is poorly understood. Endoplasmic reticulum (ER) stress has now been recognized as a pathogenic event in many chronic diseases. However, ER stress has not been studied in CP, although pancreatic acinar cells seem to be especially vulnerable to ER dysfunction because of their dependence on high ER volume and functionality. Here, we aim to investigate ER stress in CP, study its pathogenesis in relation to trypsinogen activation (widely regarded as the key event of pancreatitis), and explore its mechanism, time course, and downstream consequences during pancreatic injury. CP was induced in mice by repeated episodes of acute pancreatitis (AP) based on caerulein hyperstimulation. ER stress leads to activation of unfolded protein response components that were measured in CP and AP. We show sustained up-regulation of unfolded protein response components ATF4, CHOP, GRP78, and XBP1 in CP. Overexpression of GRP78 and ATF4 in human CP confirmed the experimental findings. We used novel trypsinogen-7 knock-out mice (T(-/-)), which lack intra-acinar trypsinogen activation, to clarify the relationship of ER stress to intra-acinar trypsinogen activation in pancreatic injury. Comparable activation of ER stress was seen in wild type and T(-/-) mice. Induction of ER stress occurred through pathologic calcium signaling very early in the course of pancreatic injury. Our results establish that ER stress is chronically activated in CP and is induced early in pancreatic injury through pathologic calcium signaling independent of trypsinogen activation. ER stress may be an important pathogenic mechanism in pancreatitis that needs to be explored in future studies.
Keywords: Chronic Pancreatitis; ER Stress; Endoplasmic Reticulum (ER); NF-kappa B (NF-KB); Pancreas; Trypsinogen Activation; Unfolded Protein Response (UPR).
© 2014 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Braganza J. M., Lee S. H., McCloy R. F., McMahon M. J. (2011) Chronic pancreatitis. Lancet 377, 1184–1197 - PubMed
-
- Etemad B., Whitcomb D. C. (2001) Chronic pancreatitis: diagnosis, classification, and new genetic developments. Gastroenterology 120, 682–707 - PubMed
-
- Guda N. M., Romagnuolo J., Freeman M. L. (2011) Recurrent and relapsing pancreatitis. Curr. Gastroenterol. Rep. 13, 140–149 - PubMed
-
- Yadav D., O'Connell M., Papachristou G. I. (2012) Natural history following the first attack of acute pancreatitis. Am. J. Gastroenterol. 107, 1096–1103 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
