

Observational study of spinal muscular atrophy type I and implications for clinical trials
- PMID: 25080519
- PMCID: PMC4155049
- DOI: 10.1212/WNL.0000000000000741
Observational study of spinal muscular atrophy type I and implications for clinical trials
Abstract
Objectives: Prospective cohort study to characterize the clinical features and course of spinal muscular atrophy type I (SMA-I).
Methods: Patients were enrolled at 3 study sites and followed for up to 36 months with serial clinical, motor function, laboratory, and electrophysiologic outcome assessments. Intervention was determined by published standard of care guidelines. Palliative care options were offered.
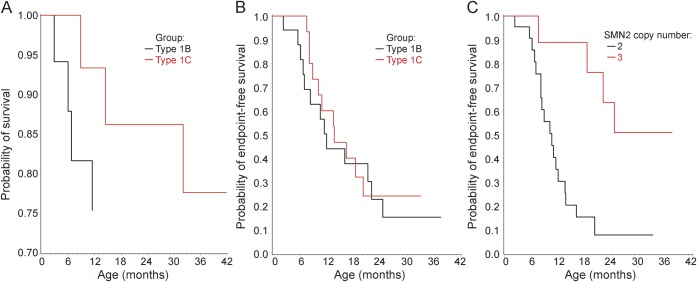
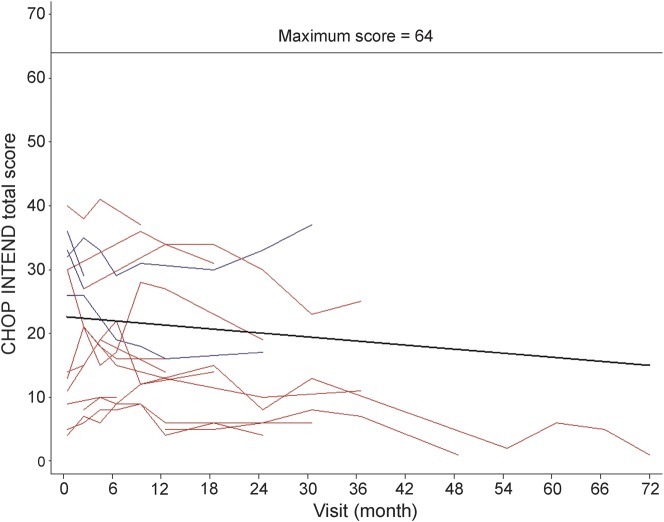
Results: Thirty-four of 54 eligible subjects with SMA-I (63%) enrolled and 50% of these completed at least 12 months of follow-up. The median age at reaching the combined endpoint of death or requiring at least 16 hours/day of ventilation support was 13.5 months (interquartile range 8.1-22.0 months). Requirement for nutritional support preceded that for ventilation support. The distribution of age at reaching the combined endpoint was similar for subjects with SMA-I who had symptom onset before 3 months and after 3 months of age (p=0.58). Having 2 SMN2 copies was associated with greater morbidity and mortality than having 3 copies. Baseline electrophysiologic measures indicated substantial motor neuron loss. By comparison, subjects with SMA-II who lost sitting ability (n=10) had higher motor function, motor unit number estimate and compound motor action potential, longer survival, and later age when feeding or ventilation support was required. The mean rate of decline in The Children's Hospital of Philadelphia Infant Test for Neuromuscular Disorders motor function scale was 1.27 points/year (95% confidence interval 0.21-2.33, p=0.02).
Conclusions: Infants with SMA-I can be effectively enrolled and retained in a 12-month natural history study until a majority reach the combined endpoint. These outcome data can be used for clinical trial design.
© 2014 American Academy of Neurology.
Figures
References
-
- Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995;80:155–165 - PubMed
-
- Gabanella F, Carissimi C, Usiello A, Pellizzoni L. The activity of the spinal muscular atrophy protein is regulated during development and cellular differentiation. Hum Mol Genet 2005;14:3629–3642 - PubMed
Publication types
MeSH terms
Grants and funding
- UL1 TR001070/TR/NCATS NIH HHS/United States
- 1 UL1 RR024156/RR/NCRR NIH HHS/United States
- R01-AR056973/AR/NIAMS NIH HHS/United States
- 5R21-NS058926/NS/NINDS NIH HHS/United States
- 1 UL1 RR025755/RR/NCRR NIH HHS/United States
- UL1 RR024134/RR/NCRR NIH HHS/United States
- K12 NS01698/NS/NINDS NIH HHS/United States
- 1R01AR060850/AR/NIAMS NIH HHS/United States
- 1U10NS077269/NS/NINDS NIH HHS/United States
- UL1 RR025755/RR/NCRR NIH HHS/United States
- U54 AR0526446-03/AR/NIAMS NIH HHS/United States
- 1U54 NS065712-01/NS/NINDS NIH HHS/United States
- 2P01NS-40828-6A11/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical