

Molecular diagnostic testing by eyeGENE: analysis of patients with hereditary retinal dystrophy phenotypes involving central vision loss
- PMID: 25082885
- PMCID: PMC4152151
- DOI: 10.1167/iovs.14-14359
Molecular diagnostic testing by eyeGENE: analysis of patients with hereditary retinal dystrophy phenotypes involving central vision loss
Abstract
Purpose: To analyze the genetic test results of probands referred to eyeGENE with a diagnosis of hereditary maculopathy.
Methods: Patients with Best macular dystrophy (BMD), Doyne honeycomb retinal dystrophy (DHRD), Sorsby fundus dystrophy (SFD), or late-onset retinal degeneration (LORD) were screened for mutations in BEST1, EFEMP1, TIMP3, and CTRP5, respectively. Patients with pattern dystrophy (PD) were screened for mutations in PRPH2, BEST1, ELOVL4, CTRP5, and ABCA4; patients with cone-rod dystrophy (CRD) were screened for mutations in CRX, ABCA4, PRPH2, ELOVL4, and the c.2513G>A p.Arg838His variant in GUCY2D. Mutation analysis was performed by dideoxy sequencing. Impact of novel variants was evaluated using the computational tool PolyPhen.
Results: Among the 213 unrelated patients, 38 had BMD, 26 DHRD, 74 PD, 8 SFD, 6 LORD, and 54 CRD; six had both PD and BMD, and one had no specific clinical diagnosis. BEST1 variants were identified in 25 BMD patients, five with novel variants of unknown significance (VUS). Among the five patients with VUS, one was diagnosed with both BMD and PD. A novel EFEMP1 variant was identified in one DHRD patient. TIMP3 novel variants were found in two SFD patients, PRPH2 variants in 14 PD patients, ABCA4 variants in four PD patients, and p.Arg838His GUCY2D mutation in six patients diagnosed with dominant CRD; one patient additionally had a CRX VUS. ABCA4 mutations were identified in 15 patients with recessive CRD.
Conclusions: Of the 213 samples, 55 patients (26%) had known causative mutations, and 13 (6%) patients had a VUS that was possibly pathogenic. Overall, selective screening for mutations in BEST1, PRPH2, and ABCA4 would likely yield the highest success rate in identifying the genetic basis for macular dystrophy phenotypes. Because of the overlap in phenotypes between BMD and PD, it would be beneficial to screen genes associated with both diseases.
Trial registration: ClinicalTrials.gov NCT00378742.
Keywords: eyeGENE; genetic testing; macular dystrophy.
Copyright 2014 The Association for Research in Vision and Ophthalmology, Inc.
Figures
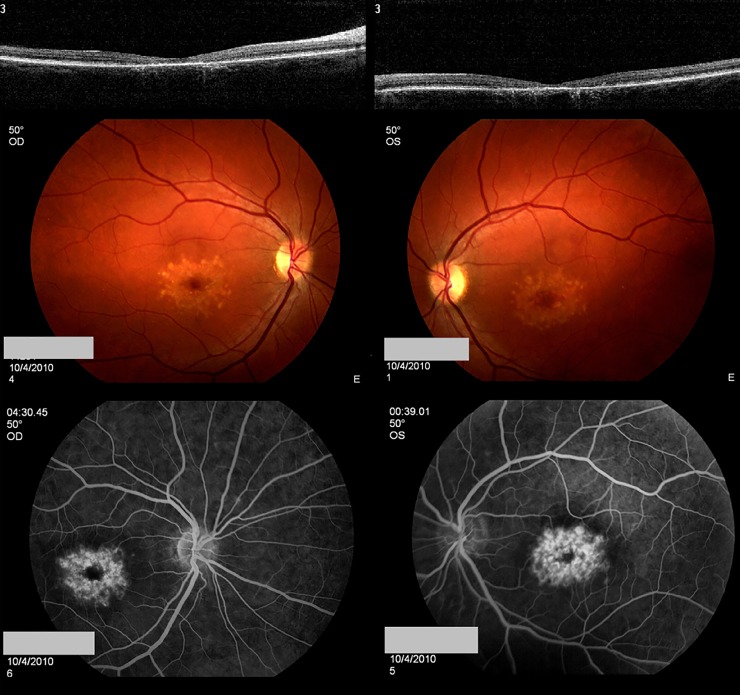
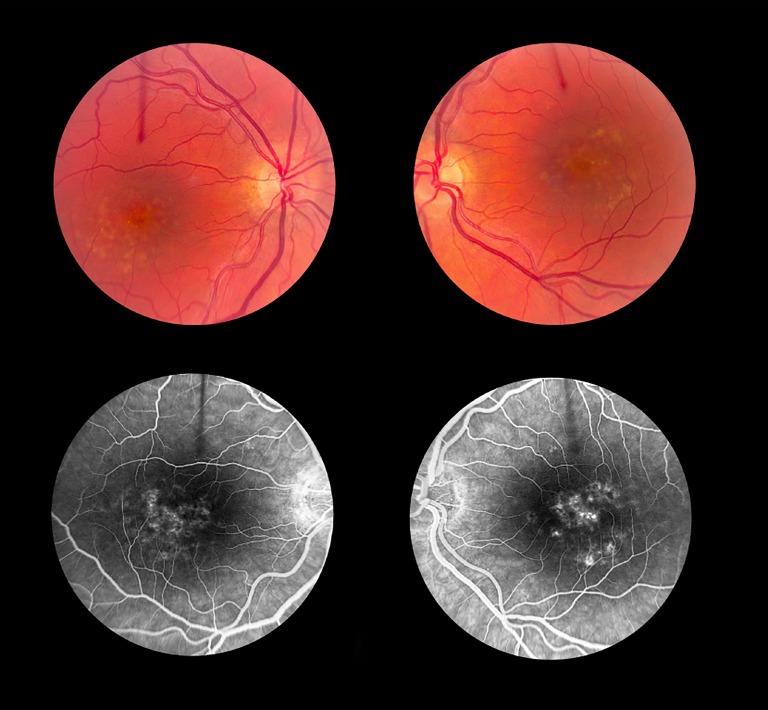
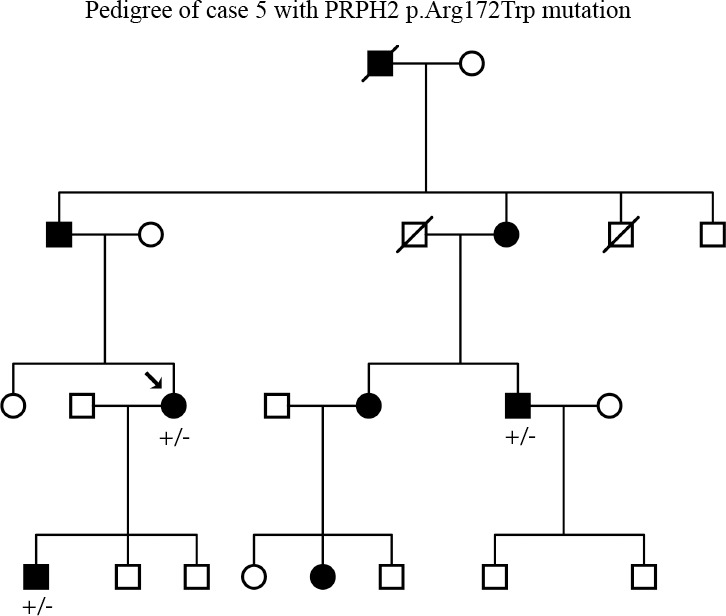
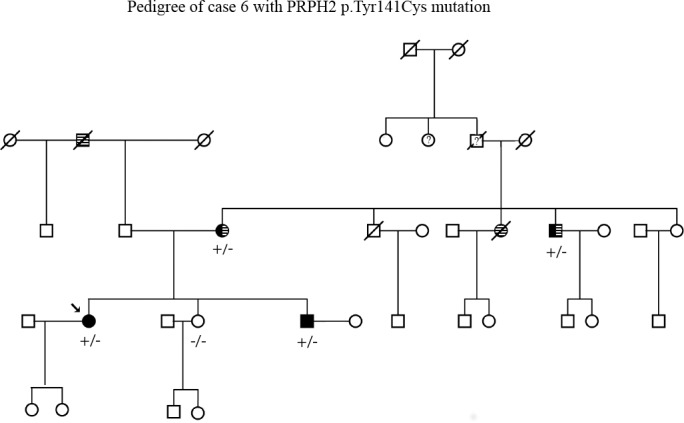
References
-
- Petrukhin K, Koisti MJ, Bakall B, et al. Identification of the gene responsible for Best macular dystrophy. Nat Genet. 1998; 19: 241–247 - PubMed
-
- Caldwell GM, Kakuk LE, Griesinger IB, et al. Bestrophin gene mutations in patients with Best vitelliform macular dystrophy. Genomics. 1999; 58: 98–101 - PubMed
-
- Stone EM, Lotery AJ, Munier FL, et al. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat Genet. 1999; 22: 199–202 - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
