

Adaptive expansion of the maize maternally expressed gene (Meg) family involves changes in expression patterns and protein secondary structures of its members
- PMID: 25084677
- PMCID: PMC4236715
- DOI: 10.1186/s12870-014-0204-8
Adaptive expansion of the maize maternally expressed gene (Meg) family involves changes in expression patterns and protein secondary structures of its members
Abstract
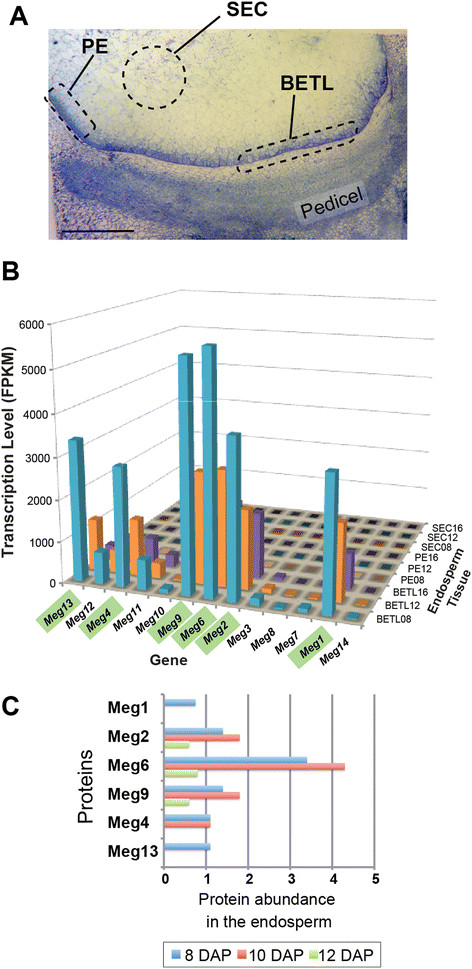
Background: The Maternally expressed gene (Meg) family is a locally-duplicated gene family of maize which encodes cysteine-rich proteins (CRPs). The founding member of the family, Meg1, is required for normal development of the basal endosperm transfer cell layer (BETL) and is involved in the allocation of maternal nutrients to growing seeds. Despite the important roles of Meg1 in maize seed development, the evolutionary history of the Meg cluster and the activities of the duplicate genes are not understood.
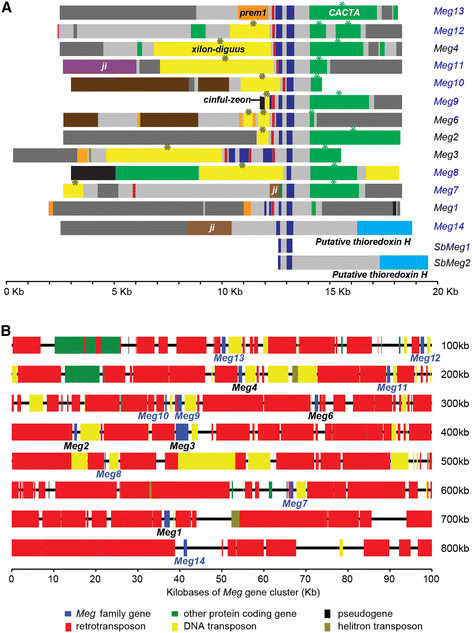
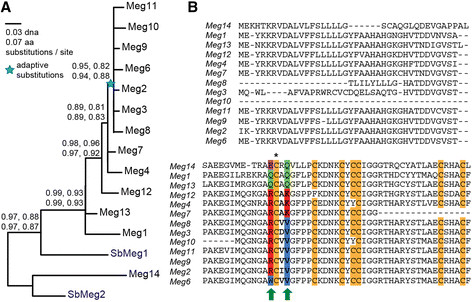
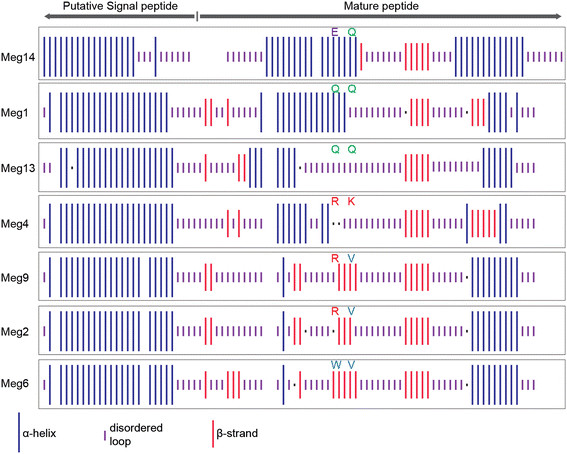
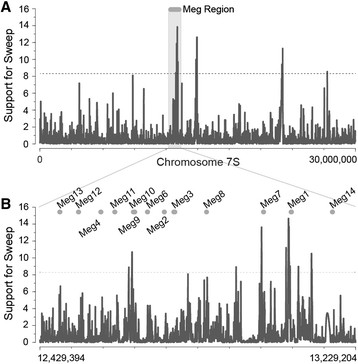
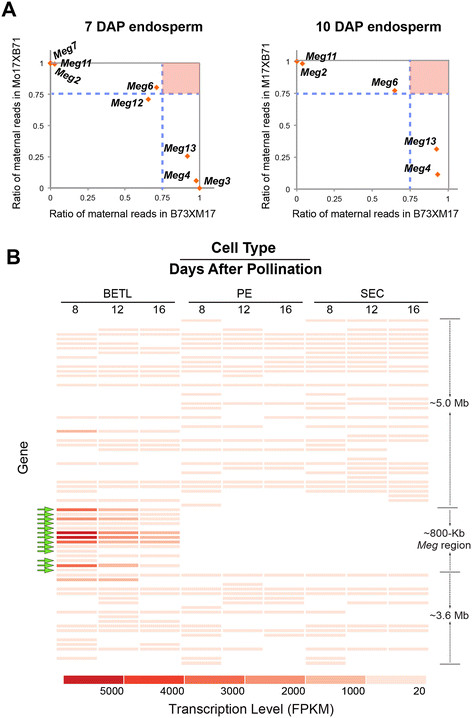
Results: In maize, the Meg gene cluster resides in a 2.3 Mb-long genomic region that exhibits many features of non-centromeric heterochromatin. Using phylogenetic reconstruction and syntenic alignments, we identified the pedigree of the Meg family, in which 11 of its 13 members arose in maize after allotetraploidization ~4.8 mya. Phylogenetic and population-genetic analyses identified possible signatures suggesting recent positive selection in Meg homologs. Structural analyses of the Meg proteins indicated potentially adaptive changes in secondary structure from α-helix to β-strand during the expansion. Transcriptomic analysis of the maize endosperm indicated that 6 Meg genes are selectively activated in the BETL, and younger Meg genes are more active than older ones. In endosperms from B73 by Mo17 reciprocal crosses, most Meg genes did not display parent-specific expression patterns.
Conclusions: Recently-duplicated Meg genes have different protein secondary structures, and their expressions in the BETL dominate over those of older members. Together with the signs of positive selections in the young Meg genes, these results suggest that the expansion of the Meg family involves potentially adaptive transitions in which new members with novel functions prevailed over older members.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
