

Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex
- PMID: 25086649
- PMCID: PMC4191988
- DOI: 10.1038/nbt.2967
Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex
Abstract
Large-scale surveys of single-cell gene expression have the potential to reveal rare cell populations and lineage relationships but require efficient methods for cell capture and mRNA sequencing. Although cellular barcoding strategies allow parallel sequencing of single cells at ultra-low depths, the limitations of shallow sequencing have not been investigated directly. By capturing 301 single cells from 11 populations using microfluidics and analyzing single-cell transcriptomes across downsampled sequencing depths, we demonstrate that shallow single-cell mRNA sequencing (~50,000 reads per cell) is sufficient for unbiased cell-type classification and biomarker identification. In the developing cortex, we identify diverse cell types, including multiple progenitor and neuronal subtypes, and we identify EGR1 and FOS as previously unreported candidate targets of Notch signaling in human but not mouse radial glia. Our strategy establishes an efficient method for unbiased analysis and comparison of cell populations from heterogeneous tissue by microfluidic single-cell capture and low-coverage sequencing of many cells.
Conflict of interest statement
A.A.L., A.P.M., B.A., B.C., B.F., B.N.J., D.K., D.T., G.S., J.A.A.W., J.S., J.W., L.K., L.S., M.A.U., M.N., M.T., M.W., N.L., N.R., P.C., R.C.J., R.L., S.W., S.W., and X.W. have a financial interest in Fluidigm Corporation as employees and/or stockholders.
Figures
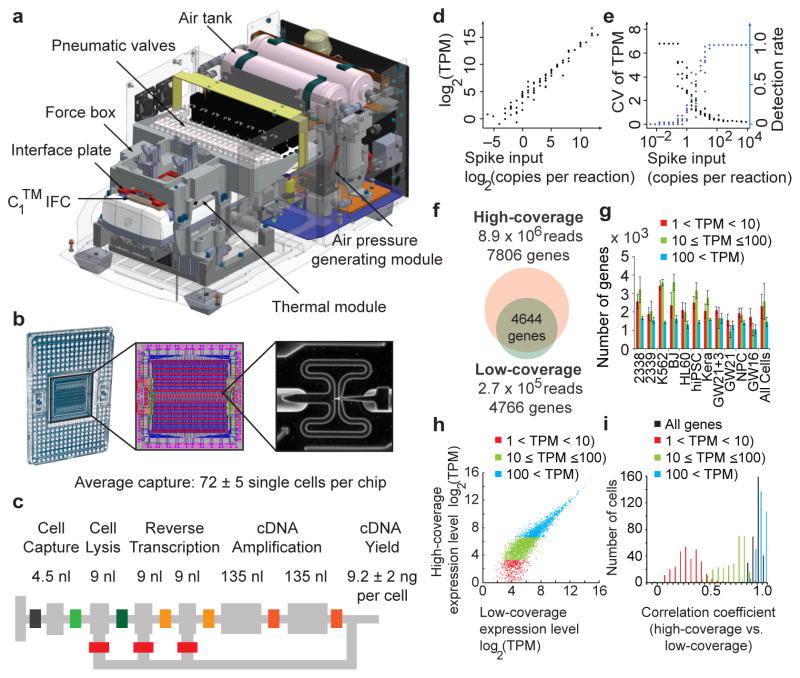
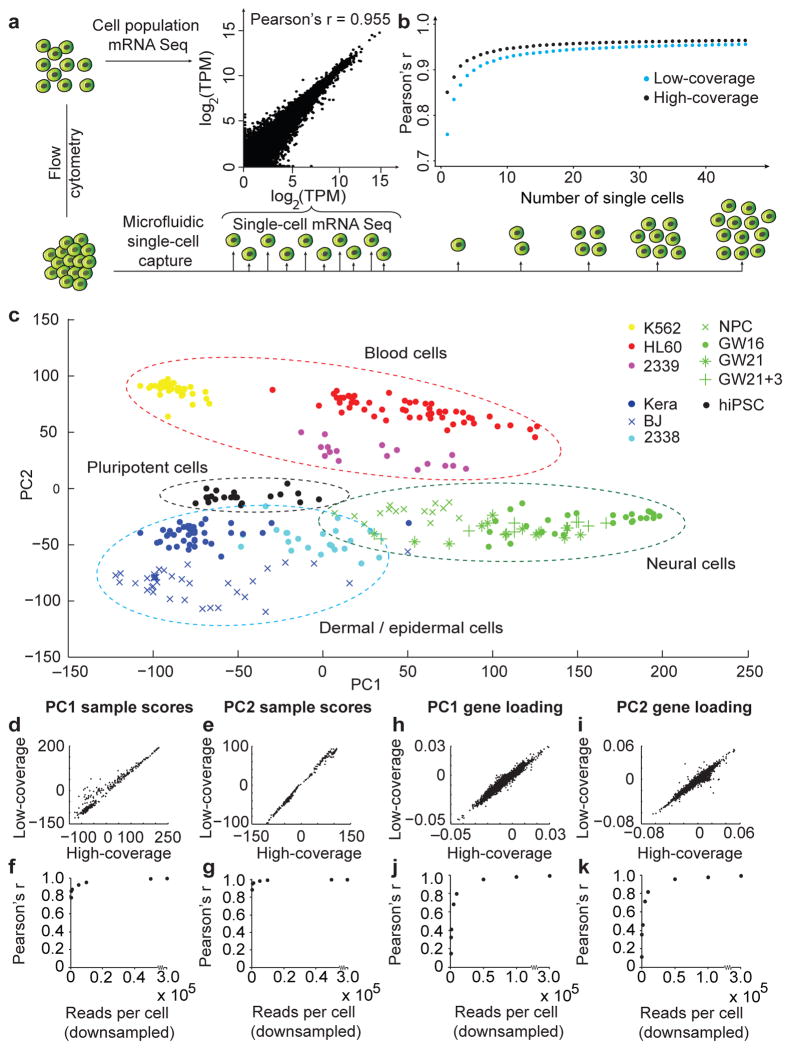
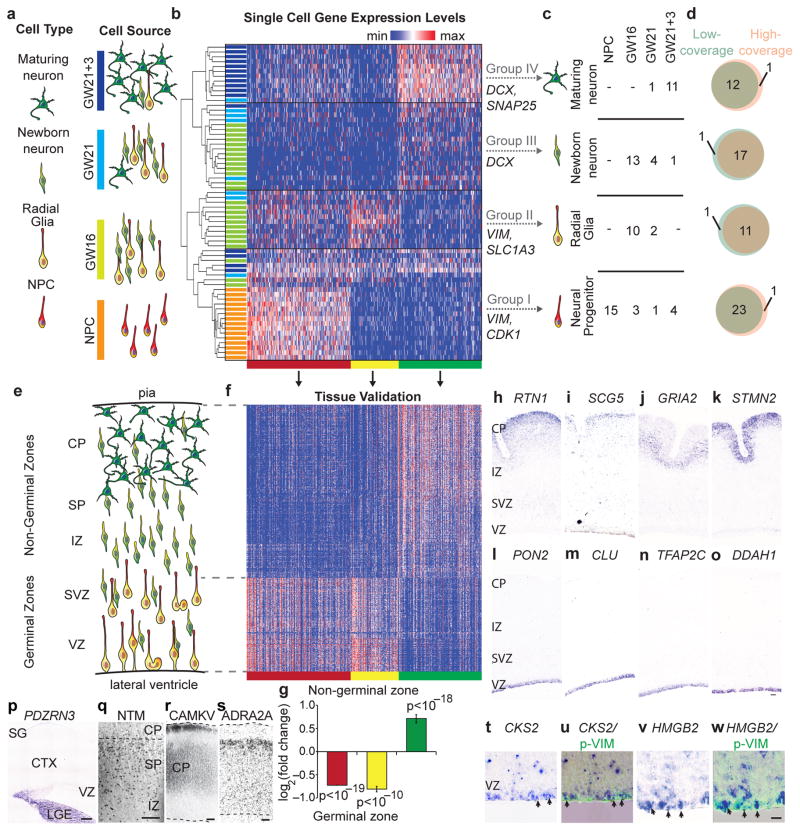
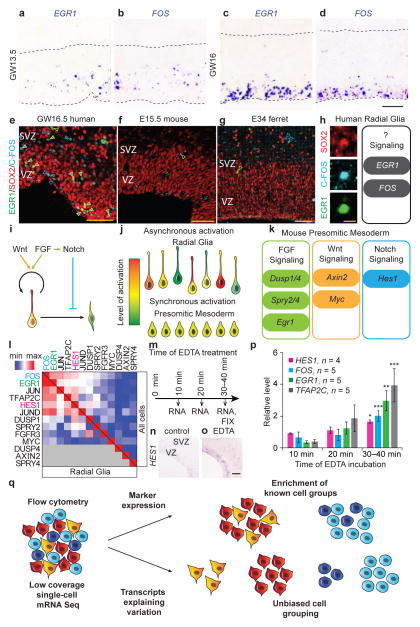
Comment in
-
How deep is enough in single-cell RNA-seq?Nat Biotechnol. 2014 Oct;32(10):1005-6. doi: 10.1038/nbt.3039. Nat Biotechnol. 2014. PMID: 25299920 No abstract available.
References
-
- Shapiro E, Biezuner T, Linnarsson S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nature reviews Genetics. 2013;14:618–630. - PubMed
-
- Kawaguchi A, et al. Single-cell gene profiling defines differential progenitor subclasses in mammalian neurogenesis. Development. 2008;135:3113–3124. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
