

Tumour heterogeneity and the evolution of polyclonal drug resistance
- PMID: 25087573
- PMCID: PMC5528620
- DOI: 10.1016/j.molonc.2014.06.005
Tumour heterogeneity and the evolution of polyclonal drug resistance
Abstract
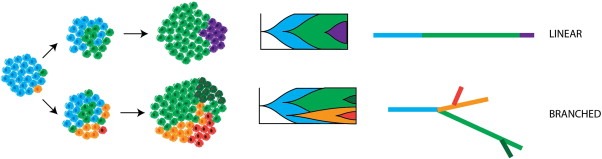
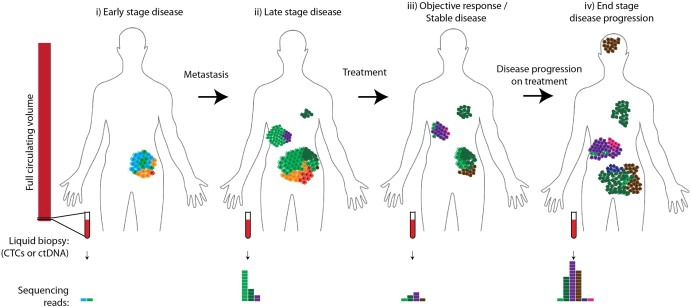
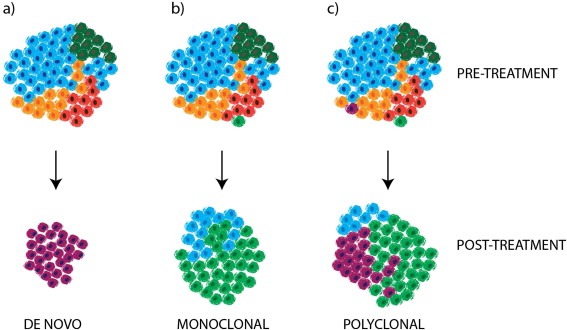
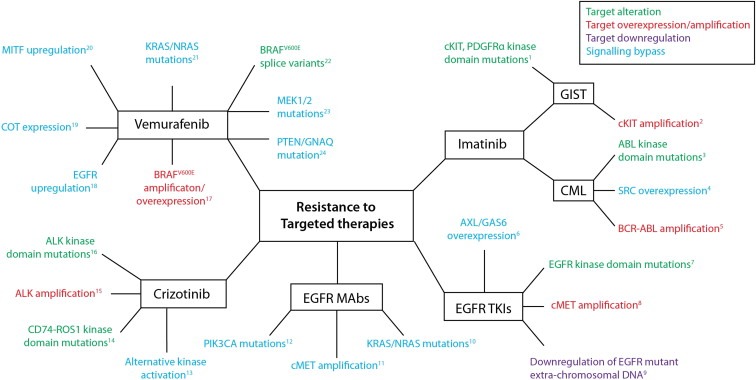
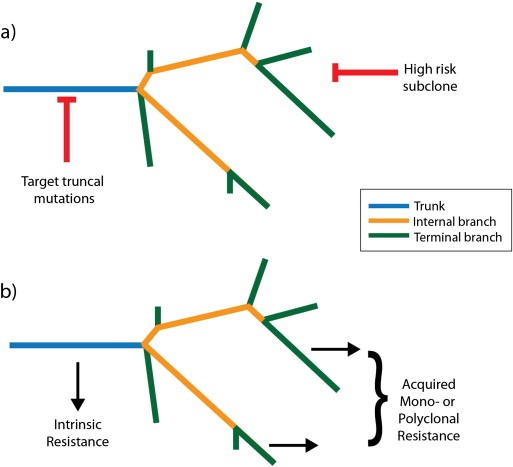
Cancer drug resistance is a major problem, with the majority of patients with metastatic disease ultimately developing multidrug resistance and succumbing to their disease. Our understanding of molecular events underpinning treatment failure has been enhanced by new genomic technologies and pre-clinical studies. Intratumour genetic heterogeneity (ITH) is a prominent contributor to therapeutic failure, and it is becoming increasingly apparent that individual tumours may achieve resistance via multiple routes simultaneously - termed polyclonal resistance. Efforts to target single resistance mechanisms to overcome therapeutic failure may therefore yield only limited success. Clinical studies with sequential analysis of tumour material are needed to enhance our understanding of inter-clonal functional relationships and tumour evolution during therapy, and to improve drug development strategies in cancer medicine.
Keywords: Cancer evolution; Drug resistance; Genomic instability; Intratumour heterogeneity.
Copyright © 2014 Federation of European Biochemical Societies. Published by Elsevier B.V. All rights reserved.
Figures
References
-
- Almendro, V. , Cheng, Y.K. , Randles, A. , Itzkovitz, S. , Marusyk, A. , Ametller, E. , Gonzalez-Farre, X. , Munoz, M. , Russnes, H.G. , Helland, A. , 2014. Inference of tumor evolution during chemotherapy by computational modeling and in situ analysis of genetic and phenotypic cellular diversity. Cell Rep.. 6, 514–527. - PMC - PubMed
-
- Amado, R.G. , Wolf, M. , Peeters, M. , Van Cutsem, E. , Siena, S. , Freeman, D.J. , Juan, T. , Sikorski, R. , Suggs, S. , Radinsky, R. , 2008. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol.: Official Journal of the American Society of Clinical Oncology. 26, 1626–1634. - PubMed
-
- Anderson, K. , Lutz, C. , van Delft, F.W. , Bateman, C.M. , Guo, Y. , Colman, S.M. , Kempski, H. , Moorman, A.V. , Titley, I. , Swansbury, J. , 2011. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 469, 356–361. - PubMed
-
- Arjan, J.A. , Visser, M. , Zeyl, C.W. , Gerrish, P.J. , Blanchard, J.L. , Lenski, R.E. , 1999. Diminishing returns from mutation supply rate in asexual populations. Science. 283, 404–406. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
