

Pathogenic variants for Mendelian and complex traits in exomes of 6,517 European and African Americans: implications for the return of incidental results
- PMID: 25087612
- PMCID: PMC4129409
- DOI: 10.1016/j.ajhg.2014.07.006
Pathogenic variants for Mendelian and complex traits in exomes of 6,517 European and African Americans: implications for the return of incidental results
Abstract
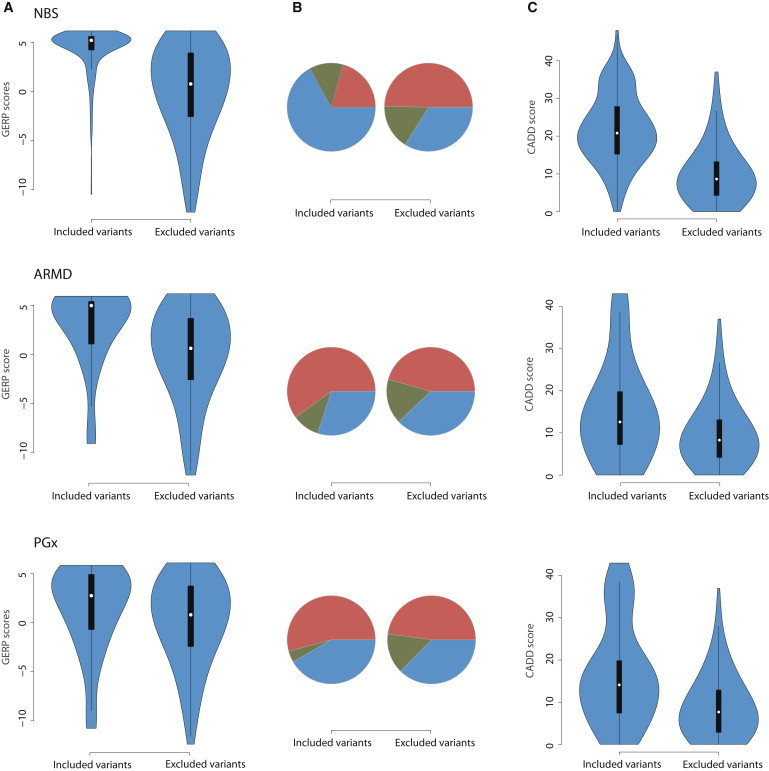
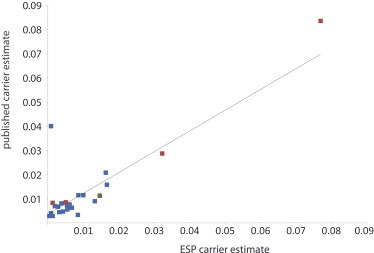
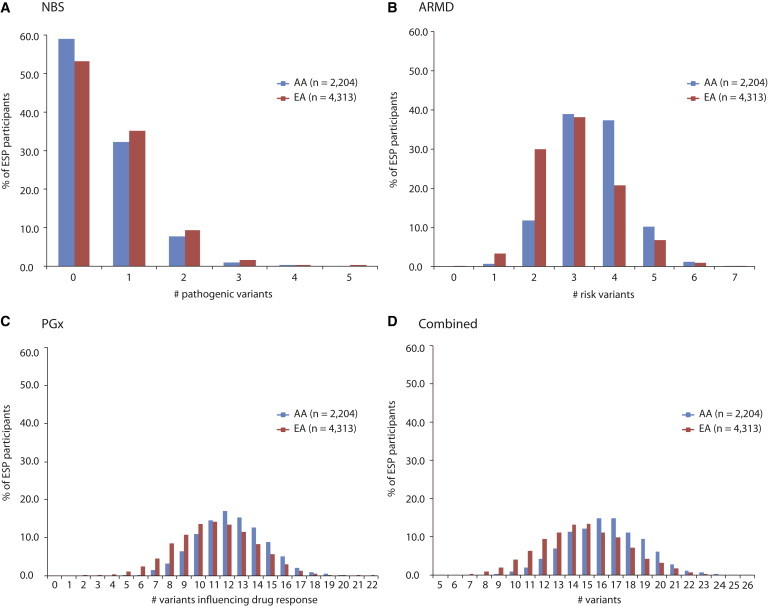
Exome sequencing (ES) is rapidly being deployed for use in clinical settings despite limited empirical data about the number and types of incidental results (with potential clinical utility) that could be offered for return to an individual. We analyzed deidentified ES data from 6,517 participants (2,204 African Americans and 4,313 European Americans) from the National Heart, Lung, and Blood Institute Exome Sequencing Project. We characterized the frequencies of pathogenic alleles in genes underlying Mendelian conditions commonly assessed by newborn-screening (NBS, n = 39) programs, genes associated with age-related macular degeneration (ARMD, n = 17), and genes known to influence drug response (PGx, n = 14). From these 70 genes, we identified 10,789 variants and curated them by manual review of OMIM, HGMD, locus-specific databases, or primary literature to a total of 399 validated pathogenic variants. The mean number of risk alleles per individual was 15.3. Every individual had at least five known PGx alleles, 99% of individuals had at least one ARMD risk allele, and 45% of individuals were carriers for at least one pathogenic NBS allele. The carrier burden for severe recessive childhood disorders was 0.57. Our results demonstrate that risk alleles of potential clinical utility for both Mendelian and complex traits are detectable in every individual. These findings highlight the necessity of developing guidelines and policies that consider the return of results to all individuals and underscore the need to develop innovative approaches and tools that enable individuals to exercise their choice about the return of incidental results.
Copyright © 2014 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Bamshad M.J., Ng S.B., Bigham A.W., Tabor H.K., Emond M.J., Nickerson D.A., Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011;12:745–755. - PubMed
-
- Below J.E., Earl D.L., Shively K.M., McMillin M.J., Smith J.D., Turner E.H., Stephan M.J., Al-Gazali L.I., Hertecant J.L., Chitayat D., University of Washington Center for Mendelian Genomics Whole-genome analysis reveals that mutations in inositol polyphosphate phosphatase-like 1 cause opsismodysplasia. Am. J. Hum. Genet. 2013;92:137–143. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
- RC2 HL102923/HL/NHLBI NIH HHS/United States
- RC2 HL102926/HL/NHLBI NIH HHS/United States
- RC2 HL-102926/HL/NHLBI NIH HHS/United States
- RC2 HL-102923/HL/NHLBI NIH HHS/United States
- T32 HL007208/HL/NHLBI NIH HHS/United States
- P20 MD006899/MD/NIMHD NIH HHS/United States
- RC2 HL-102925/HL/NHLBI NIH HHS/United States
- RC2 HL103010/HL/NHLBI NIH HHS/United States
- U01 AG049505/AG/NIA NIH HHS/United States
- RC2 HL-102924/HL/NHLBI NIH HHS/United States
- RC2 HL102924/HL/NHLBI NIH HHS/United States
- RC2 HL-103010/HL/NHLBI NIH HHS/United States
- RC2 HL102925/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
