

SPEG interacts with myotubularin, and its deficiency causes centronuclear myopathy with dilated cardiomyopathy
- PMID: 25087613
- PMCID: PMC4129406
- DOI: 10.1016/j.ajhg.2014.07.004
SPEG interacts with myotubularin, and its deficiency causes centronuclear myopathy with dilated cardiomyopathy
Abstract
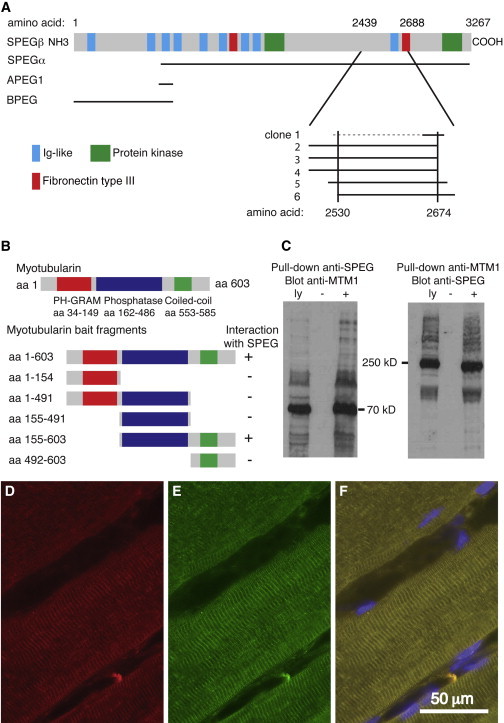
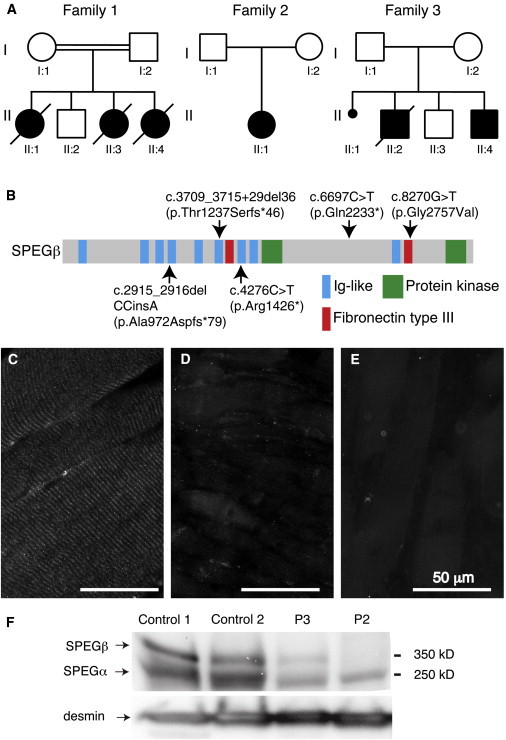
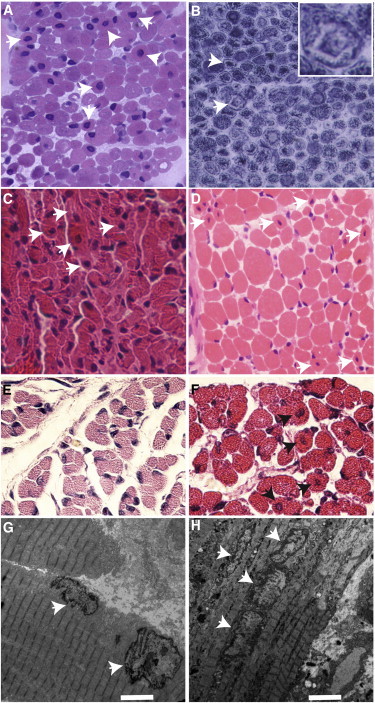
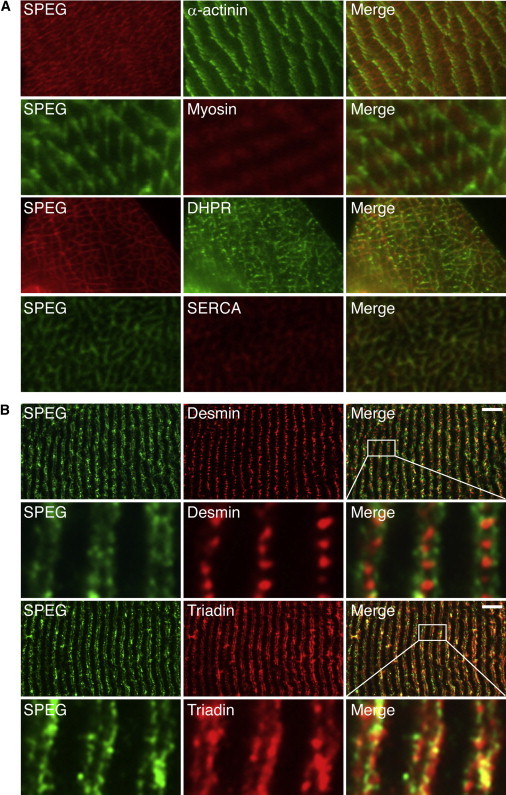
Centronuclear myopathies (CNMs) are characterized by muscle weakness and increased numbers of central nuclei within myofibers. X-linked myotubular myopathy, the most common severe form of CNM, is caused by mutations in MTM1, encoding myotubularin (MTM1), a lipid phosphatase. To increase our understanding of MTM1 function, we conducted a yeast two-hybrid screen to identify MTM1-interacting proteins. Striated muscle preferentially expressed protein kinase (SPEG), the product of SPEG complex locus (SPEG), was identified as an MTM1-interacting protein, confirmed by immunoprecipitation and immunofluorescence studies. SPEG knockout has been previously associated with severe dilated cardiomyopathy in a mouse model. Using whole-exome sequencing, we identified three unrelated CNM-affected probands, including two with documented dilated cardiomyopathy, carrying homozygous or compound-heterozygous SPEG mutations. SPEG was markedly reduced or absent in two individuals whose muscle was available for immunofluorescence and immunoblot studies. Examination of muscle samples from Speg-knockout mice revealed an increased frequency of central nuclei, as seen in human subjects. SPEG localizes in a double line, flanking desmin over the Z lines, and is apparently in alignment with the terminal cisternae of the sarcoplasmic reticulum. Examination of human and murine MTM1-deficient muscles revealed similar abnormalities in staining patterns for both desmin and SPEG. Our results suggest that mutations in SPEG, encoding SPEG, cause a CNM phenotype as a result of its interaction with MTM1. SPEG is present in cardiac muscle, where it plays a critical role; therefore, individuals with SPEG mutations additionally present with dilated cardiomyopathy.
Copyright © 2014 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Pierson C.R., Tomczak K., Agrawal P., Moghadaszadeh B., Beggs A.H. X-linked myotubular and centronuclear myopathies. J. Neuropathol. Exp. Neurol. 2005;64:555–564. - PubMed
-
- Romero N.B., Bitoun M. Centronuclear myopathies. Semin. Pediatr. Neurol. 2011;18:250–256. - PubMed
-
- Laporte J., Hu L.J., Kretz C., Mandel J.L., Kioschis P., Coy J.F., Klauck S.M., Poustka A., Dahl N. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat. Genet. 1996;13:175–182. - PubMed
-
- Bitoun M., Maugenre S., Jeannet P.Y., Lacène E., Ferrer X., Laforêt P., Martin J.J., Laporte J., Lochmüller H., Beggs A.H. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat. Genet. 2005;37:1207–1209. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
