

Molecular mechanisms and animal models of spinal muscular atrophy
- PMID: 25088406
- PMCID: PMC12184990
- DOI: 10.1016/j.bbadis.2014.07.024
Molecular mechanisms and animal models of spinal muscular atrophy
Abstract
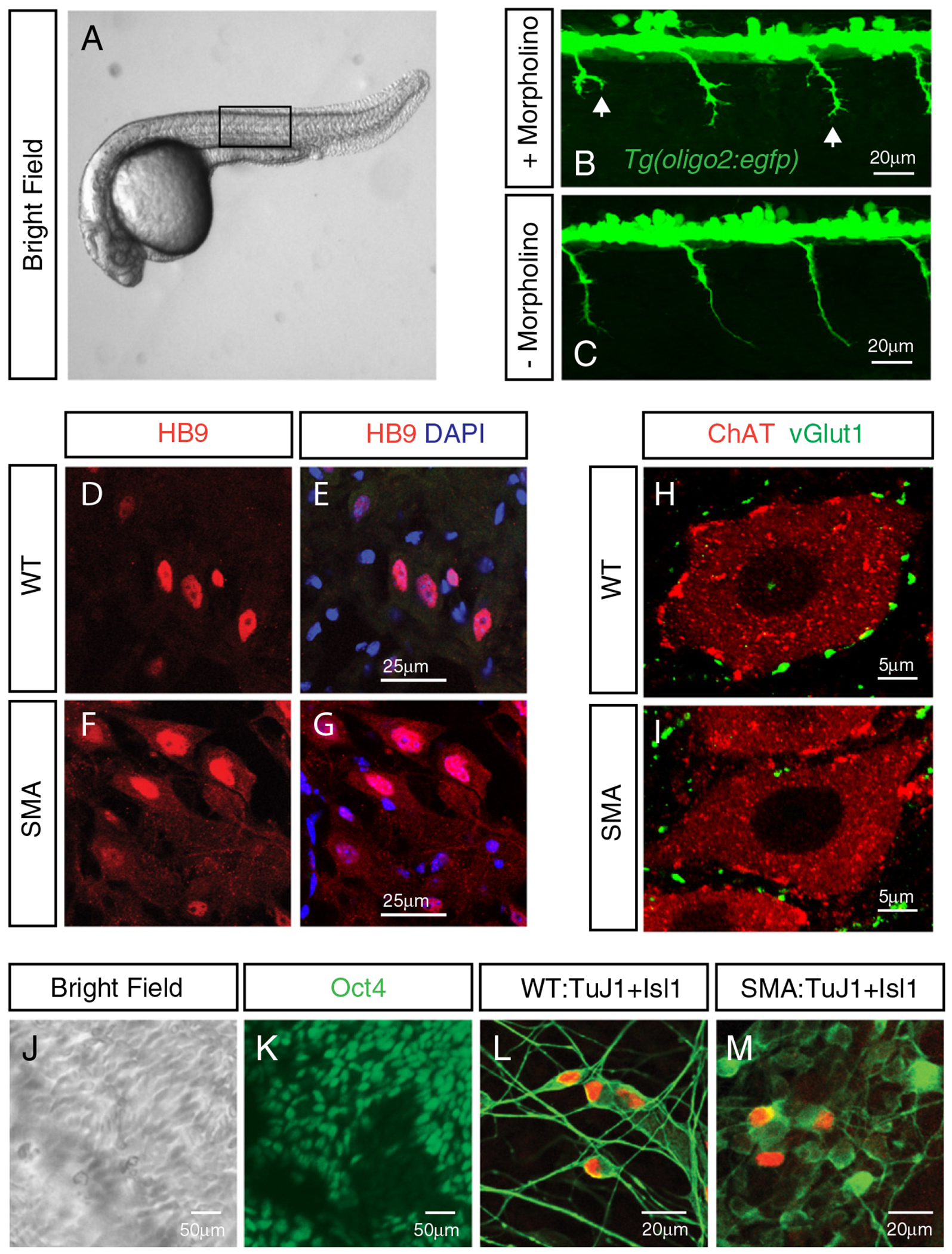
Spinal muscular atrophy (SMA), the leading genetic cause of infant mortality, is characterized by the degeneration of spinal motor neurons and muscle atrophy. Although the genetic cause of SMA has been mapped to the Survival Motor Neuron1 (SMN1) gene, mechanisms underlying selective motor neuron degeneration in SMA remain largely unknown. Here we review the latest developments and our current understanding of the molecular mechanisms underlying SMA pathogenesis, focusing on the animal model systems that have been developed, as well as new diagnostic and treatment strategies that have been identified using these model systems. This article is part of a special issue entitled: Neuromuscular Diseases: Pathology and Molecular Pathogenesis.
Keywords: Animal disease models; C. elegans; Drosophila; SMA; SMN; Zebrafish.
Copyright © 2014 Elsevier B.V. All rights reserved.
Figures

References
-
- Andreassi C, Angelozzi C, Tiziano FD, Vitali T, De Vincenzi E, Boninsegna A, Villanova M, Bertini E, Pini A, Neri G, et al. , Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy, Eur. J. Hum. Genet 12 (2004) 59–65. - PubMed
-
- Araki S, Hayashi M, Tamagawa K, Saito M, Kato S, Komori T, Sakakihara Y, Mizutani T, Oda M, Neuropathological analysis in spinal muscular atrophy type II, Acta Neuropathol. 106 (2003) 441–448. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
